北京科音量子化学波函数分析与Multiwfn程序培训班
下一届本培训举办时间见北京科音首页http://www.keinsci.com的预告栏。着急开展研究、等不及下届培训者可以购买往届培训资料自学,方式见本页面购买往届资料部分。仔细阅读本页面以及《北京科音办的培训班FAQ》后若对培训仍有不清楚的地方请发邮件至北京科音官方邮箱keinsci@sina.com咨询,我们会尽快回复。
本页面最后更新:2026-Apr-27
培训介绍
电子的波函数分析是量子化学研究实际化学问题的几乎必不可少的手段。如果你觉得写量子化学应用性研究文章时没什么可分析和讨论的内容,说明你最缺乏的就是波函数分析知识!若能掌握波函数分析知识、充分运用波函数分析手段,在量子化学研究中就会如鱼得水、如虎添翼,分析视角将变得十分广阔,很容易将问题研究得深入、全面、透彻,并从而写出内容充实的优秀文章,最终发表在高档次刊物上。例如北京科音的卢天等人在碳化学权威刊物Carbon发表的四篇关于18碳环的量子化学研究工作,以及在知名的Acc. Mater. Res.、Chem. Eur. J、Chem. Commun.、Chem Eng. J.、Inorg. Chem.等期刊上发表的诸多相关工作(汇总见http://sobereva.com/carbon_ring.html),就极大程度依赖于波函数分析来对现象和本质进行深入的剖析、获得新颖独特的视角,充分体现了波函数分析与Multiwfn程序在实际研究中的关键性价值。
为什么学习波函数分析是非常重要的而不是可有可无的?如果不懂波函数分析,量子化学研究时就会感到分析手段非常局限,没法把问题研究得深入、透彻,文章会显得非常空洞,就如同吃饭时只有主食而没有任何菜肴,也自然难以发表到高档次期刊上。比如研究电子激发问题,倘若不掌握波函数分析方法,就只能算算激发能、振子强度、画个UV-Vis谱、看看分子轨道,显得非常肤浅、千篇一律,多么套路和乏味、毫无新意和亮点!而实际上,在波函数分析领域,有巨量电子激发分析方法可用,比如空穴-电子分析、DCT指数、Sr指数、电荷转移光谱分析、密度差分析、激发态成键相对于基态的变化分析、NTO分析、跃迁密度矩阵、跃迁偶极矩密度分析,等等等等。掌握这些分析并充分运用在研究和讨论中,无疑会令研究工作极大地增光添彩!再比如,你是主要研究的是分子间弱相互作用问题,如果不懂波函数分析,就只能优化一下复合物的几何结构、算一下结合能、看看振动频率和模式,根本什么深层本质的也了解不了。但如果你掌握了Multiwfn提供的弱相互作用分析的巨型“波函数分析武器库”,那分析起来将得心应手,能把相互作用的本质考察得十分透彻,从而充分解释当前研究的相互作用,并归纳出规律预测类似的相互作用的特征,获得发表文章时审稿人最看重的physical insight!
北京科音自然科学研究中心(http://www.keinsci.com)的卢天开发的Multiwfn(http://sobereva.com/multiwfn)是功能非常强大、十分易于上手和使用、效率很高、开源免费、自主开发的量子化学波函数分析程序,它使得各种波函数分析方法都可以非常方便地应用到各种实际问题的研究中,在世界范围已经产生了很大影响力。Multiwfn从2009年至今一直在快速发展,用户现已遍及世界超过90个国家,已被多达超过40000篇高水平计算化学研究论文或专著所使用!其中包括Science、Nature、Nature Chemistry/Materials/Energy/Photonics/Physics/Catalysis/Synthesis/Sustainability/Nanotechnology、JACS、Angew、PNAS、Adv. Mater.等大量国际顶级刊物,以及Truhlar、Perdew、Grimme、Morokuma、Frank Neese、Houk、Shaik等著名量子化学家的研究文章。Multiwfn程序的基本常识和相关介绍见Multiwfn FAQ和Multiwfn入门tips。
北京科音举办的量子化学波函数分析与Multiwfn程序培训班会对波函数分析方法的原理和应用进行非常全面和深入的介绍,完整、系统地讲授强大的Multiwfn波函数分析程序的使用方法,以使参加者们充分地拓展视野、明显提升量子化学研究水准和文章档次。NBO、AICD等一些其它与波函数分析范畴有关的程序也会在培训中详细讲解。本培训始终强调将理论、程序操作与实际问题紧密结合,令学员们能够学以致用。本培训干货满满,我们相信做过一定量子化学研究的学员们在参加过本培训后肯定会感到如获至宝。本培训对学员的量子化学的基础要求不高,只要会通过Gaussian或其它主流量子化学程序做单点能、几何优化等基本的量子化学计算即可顺利学习本培训的内容。
Multiwfn不仅可以对分子、团簇这样的孤立体系进行分析,也支持对周期性体系进行分析,这在本培训里也有讲授。因此Multiwfn做波函数分析的应用绝不仅限于量子化学研究范畴,对于第一性原理研究固体、表面等周期性体系也极其重要!
特别值得强调的是,本培训的内容极为深入详尽,且注重深入浅出,构成了十分完备的知识体系。之所以本培训的幻灯片能有多达超过2600页的规模,是因为讲授的十分浩瀚,而且大部分内容都是在公开的博文/帖子里根本没有的。哪怕是已经通读过sobereva写的所有波函数分析相关博文/帖子且完整阅读过Multiwfn手册的Multiwfn老用户,参加本培训后在波函数分析的理论和应用方面的水平也会有极大的提升!看零碎的博文/帖子能获取到的知识和参加本培训所学到的知识,在广度、深度和系统性上都有天壤之别。
本培训由于很高的含金量、非常充实的内容以及良心的价格,得到历届参加者们的高度好评!往届培训回顾和学员们的评论见http://bbs.keinsci.com/forum.php?mod=forumdisplay&fid=43&filter=typeid&typeid=247。第8届培训的学员评论摘录:




第9届培训的学员评论摘录:



第10届培训的学员评论摘录:
培训费用
每一届培训费用会酌情调整,目前培训费用如下。自费比公费便宜很多是为了尽可能减少自掏腰包学习的学员的经济压力。如果参加过以前任意一届本培训,若再次参加以学习新加入的知识,将享受7折优惠价。
• 学生自费价:1600元
• 教师(含博士后)以及企业人员自费价:1900元
• 学生公费价:2300元
• 教师(含博士后)公费价:2500元
• 企业人员公费价:2800元
购买往届资料(等同于不到现场参加。不叫线上培训):上一届培训的纸质讲义(不提供电子版ppt)、现场录音(没有现场录像)、电子资料(培训中涉及的共1000个以上文件、相关程序、约200个重要的原文/综述/教程)、补充视频(仅包含前述的现场来不及讲的部分)可以在任何时间向北京科音官方购买。纸质讲义通过快递发送(支持顺丰寄往海外,邮费自理),电子资料通过网盘提供。由于讲义和讲授音频都极其详细,通过这些资料自学也有很好的效果(如果对不到场学习效果有顾虑的话可以参考以往不到场学员在此帖里的评论)。如果有自学时有没搞懂的,可以在非常知名和专业的计算化学交流群思想家公社QQ群里问,卢老师(群主+讲授者)都会非常及时、耐心解答。有购买需求者可随时发邮件至北京科音官方邮箱keinsci@sina.com与我们联系,邮件里请注明要购买的培训班的名称,费用和到现场参加往届培训相同(如果学员是自费参加,并且之前使用过Multiwfn程序发表过论文,费用可减免200元。在邮件中请注明之前发表过的文章)。可以开具发票以供报销,可提供邀请函。(若2日内未收到回复邮件,请注意检查是否被误归入了垃圾邮件分类,如果里面仍没找到,可以换个邮箱再发)
“量子化学计算+分析”优惠套装: 量子化学波函数分析与Multiwfn程序培训班 + 初级量子化学培训班,一次性购买两门课的往届资料并同时付款可享受200元优惠,两门课都学完后又会量子化学计算又掌握了海量的分析方法,做出内容丰富、富有深度的研究就不再困难!(邮件里请主动注明要以套装价购买)
郑重声明:本培训的讲义、录音、电子资料皆严禁擅自复制、传播、倒卖。我们会经常对互联网进行搜索,以及钓鱼方式收集证据。在计算化学领域的诸多学术QQ群里、公众号、频道up主的订阅者里也都有大量北京科音往届的学员,当他们发现非法利用培训资料的行为时都会向我们举报。每本讲义在印刷时都加入了大量隐藏标记,并且在发放时做了登记,我们根据讲义内容可以直接查出来领取的学员姓名和联系方式。非法利用我们的培训资料的行为一经发现,必将严厉对涉事者追究法律责任、要求赔偿北京科音的经济损失、通报相关学校/单位。同时敬告某些人,切勿试图通过非正规途径获得本课程资源,否则一定会被坑,得到的会是过时版本(北京科音培训的讲义和相关电子文件更新得很频繁)、残缺不全的资料、文字和图片模糊不清,等于花钱白打水漂,而且还无法通过身份核验而在思想家公社QQ群里成为VIP成员,因此得不到本培训讲授者卢天老师的专业、详细、耐心的答疑(答疑内容包括但不限于课上讲授的)。
培训内容
注:以下没有专门提及的信息在《北京科音办的培训班FAQ》(http://www.keinsci.com/FAQ.html)里基本都能找到答案。
本培训讲授者为北京科音自然科学研究中心主任卢天,是Multiwfn程序的开发者,长期致力于波函数分析理论和程序的发展,并对量子化学和分子模拟有深入研究,个人介绍见北京科音官网的“人才队伍”页面。
本培训的信息量巨大,精心制作的幻灯片多达超过2600页!!!培训为时五天(共5*8=40小时。答疑时间另计),由于内容实在太丰富,以尽可能高效率在现场也没法完全讲完,因此还提供约14个小时的补充视频,相当于一天半的授课量,因此本培训是5+1.5天。
培训有专属的QQ群,现场培训结束后两周内如果对培训所讲的内容有任何疑问、没完全搞懂的地方都可以尽管在群里提问。即便在群解散后,我们依然会在精力允许的情况下无限期、无偿地在总计超过一万人的规模巨大、十分活跃的综合性计算化学QQ群“思想家公社”和高度专业、人气特别高的计算化学论坛“计算化学公社”中对学员学习培训内容和科研过程中遇到的问题进行解答。学员的重要专属福利:参加本培训的学员,在思想家公社QQ群里经管理员核验报名信息后,可以成为永久VIP成员,讲授者(即群主Sobereva)在回复VIP成员的提问时会远比回复普通群成员的优先级高得多、具体得多!详见公社QQ群VIP制度。
Multiwfn的发展极快,每届培训内容都会有明显的更新和扩充,往届学员参加下一届都能享受高折扣优惠价。
静电势(ESP)、范德华势、ALIE/LEAE、键级及其分解、原子电荷与布居分析、轨道成份、DOS、COHP、Bader的QTAIM、ELF、LOL、IGMH、IRI、ETS-NOCV、NAdO、能量分解、CDA、氧化态、电子密度/自旋密度、能量密度、电子密度差、STM、NBO/NPA/NAO/NLMO/NRT、E2超共轭、AICD、GIMIC、NICS、ICSS、AdNDP、概念密度泛函理论(福井函数/双描述符/亲电与亲核指数/软度/化学势/电负性...)、空穴-电子分析、NTO等等各种波函数分析方法及一切相关背景知识全都会在本培训里做最为完整全面的介绍!很多概念和方法你之前没听说过,而在你学完了本培训之后就会意识到是多么重要!本培训的内容和讲授顺序如下,各主题的ppt页数一并列出。除以下罗列的内容外培训中还提供大量精心准备的练习,做过之后不仅能更好掌握程序使用,还会扩展视野、启迪思维。
- 波函数分析的预备知识(30多页):介绍波函数分析的基本含义、意义、用处、内容。介绍与波函数分析有关的量子化学基础知识,包括各种轨道的定义和本质、量子化学用的计算形式、基函数、电子密度、电子对密度、密度矩阵等。
- Multiwfn概览(约40页):介绍Multiwfn的特色、优点、对周期性的考虑、开发历史、现状、发展方向、求助方式、相关学习资源。介绍Multiwfn的界面特点、计算效率、支持的格式、功能选项的层次、支持的实空间函数、主要用途等。
- Multiwfn的使用基础(80多页):详细介绍手册编排、对不同平台支持情况、程序文件和运行参数设定、不同平台下的安装配置、控制输出图像格式和尺寸的方法、基本使用流程。然后依次详细介绍Multiwfn支持的各种文件类型,说明Multiwfn计算分析需要的信息和输入文件类型的选取问题。讲解一些注意事项,比如赝势下做波函数分析、给Multiwfn提供晶胞信息、如何检验波函数是否正确载入了、如何在比CCSD更高理论级别下做波函数分析。之后介绍怎么通过各种量子化学和第一性原理程序程序产生Multiwfn需要的输入文件、怎么用Multiwfn转化文件格式等等。最后介绍使用技巧:使窗口能够记录更多输出、从屏幕上拷贝数据、以纯命令方式运行、快速载入文件。
- 使用Multiwfn绘制轨道(30多页):详细、全面讲解怎么使用Multiwfn绘制分子轨道、查看轨道信息、调节图像效果等,使学员充分熟悉Multiwfn的等值面显示界面,并能根据需要轻车熟路地调节效果做出非常好的图。同时介绍怎么显示轨道中指定原子贡献的部分、怎么与VMD结合非常方便地绘制出极其出色的分子轨道图像和多层叠加的效果、妙用AI结合Multiwfn画的原图快速得到能上期刊封面的惊艳的轨道图像、用AI结合提示词后期轻松调节Multiwfn产生的轨道图像效果
- NBO方法的原理与NBO程序使用基础(约170页):这一部分首先深入讲解NBO方法的框架、基本思想,然后结合实际体系依次详细介绍NAO、NBO、NHO、NLMO、E2分析等概念。之后介绍NBO理论的局限性、如何正确看待NBO的输出。接下来讲Weinhold开发的官方NBO程序,包括如何独立运行和结合不同量化程序运行,如何使用关键词,并说明NBO分析时计算级别的选择问题。之后结合具体分子(闭壳层体系、开壳层体系、强共振体系、含多种心作用体系、环张力体系等)详细全面解读NBO输出的各种信息,令学员搞明白怎么用NBO程序分析实际体系来得到有用的信息,从而能够分析和解释化学问题。之后介绍NBO搜索方面的高级知识,包括$CHOOSE定向搜索、Hyperbond搜索等。然后解释NBO相位问题、介绍如何考察原子轨道杂化导致的能量变化、介绍将NBO给出的Lewis结构通过MarvinView直接绘制的方法。之后详细介绍自然共振理论(NRT)分析,包括原理、算法细节、程序用法等。最后介绍如何通过Multiwfn(包括结合VMD)绘制NBO产生的各种轨道图形。
- 轨道成分分析(约50页):介绍轨道成分分析的意义和用途,介绍各种轨道成份计算方法(Mulliken方法、SCPA等修改的Mulliken方法、AOIM方法、NAO方法、Hirshfeld/Becke方法等),对方法选用提供建议。然后示例用Multiwfn通过不同轨道成份计算方法分析不同体系的轨道成份(基函数贡献、原子轨道贡献、原子贡献、片段贡献)。然后介绍通过轨道离域化指数(ODI)分析轨道空间离域程度。之后介绍在NBO程序里通过CMO关键词考察分子轨道中各个NBO轨道所占成份的方法。最后介绍Multiwfn中分析不同来源的轨道之间的对应性关系的功能的使用。
- 绘制分子的态密度(DOS)、COHP、光电子谱和模拟扫描隧道显微镜(STM)图像(约80页):介绍总态密度(TDOS)、部分DOS (PDOS)、重叠DOS (OPDOS)、分子轨道PDOS (MO-PDOS)的概念和实际应用。深入讲解如何在Multiwfn中做DOS分析,如何通过调整各种选项实现研究目的和达到理想图像效果,并给出有代表性的分析操作例子。还介绍d-band center和局部DOS (local DOS)的概念、意义以及在Multiwfn中的绘制方式。之后介绍如何用DOS模块的光电子谱绘制界面来非常简单且可靠地基于Generalized Koopmans定理绘制光电子谱。介绍分析化学键很流行的COHP的概念、使用Multiwfn绘制COHP图以及计算其积分的方法。最后介绍如何模拟隧道扫描显微镜(STM)图像。
- 布居分析与原子电荷(140多页):介绍布居分析与原子电荷的基本概念和价值,对几十种计算方法进行归类,然后依次介绍算法和思想,包括Mulliken、修改的Mulliken、Lowdin、拟合静电势方法(CHELPG、MK、RESP、RESP2、REPEAT)、AIM、Voronoi、Hirshfeld、Hirshfeld-I、MBIS、cMBIS、DDEC、CM5、GAPT、AM1-BCC、MMFF94、EEM、QEq、PEOE等方法,以及培训讲授者卢老师提出的非常流行的ADCH方法,以及其第三方变体ADCHα-I。之后对不同原子电荷计算方法进行系统性的对比讨论,并对方法选用进行推荐。之后,结合各种体系(包括周期性体系)和各种实际问题,示例如何通过Multiwfn使用各种方法做布居分析和原子电荷计算来解决各种实际问题,其中重点展现Multiwfn在计算RESP电荷上的强大和灵活。之后介绍自旋布居的概念、计算和分析方法,以及自旋自然轨道的产生和分析。最后还介绍特殊技巧,包括利用布居分析判断基函数与原子轨道对应关系、根据原子属性进行着色。
- 键级分析(70多页):介绍键级的基本概念,然后介绍各种键级的定义、特点和思想,包括形式键级、分子轨道理论定义的键级、自然共振理论键级、NLMO键级、AIM键级、Coulson和Mulliken键级、Wiberg键级(及其分解为NAO原子轨道对贡献的方法)、Mayer键级、广义化的Wiberg键级、Atomic-orbital-symmetry based (AOSB)分解方法、轨道占据数扰动的Mayer键级、多中心键级、离域化指数、模糊键级,以及卢老师提出的拉普拉斯键级(LBO)。然后通过大量实际体系和不同问题对比不同键级的特点和优缺点,并说明键级计算适合的计算级别。之后,通过丰富、种类多样的实例演示如何通过Multiwfn计算各种类型的键级研究实际问题,其中还包括周期性体系。最后还说明怎么在NBO程序中利用bndidx等关键词计算键级。
- ETS-NOCV分析(30多页):介绍ETS-NOCV方法的思想、原理、算法、应用,如何通过Multiwfn实现ETS-NOCV分析,并给出诸多实例展现出ETS-NOCV在分析片段间轨道相互作用强度和本质方面的不可替代的极为重要的用处。
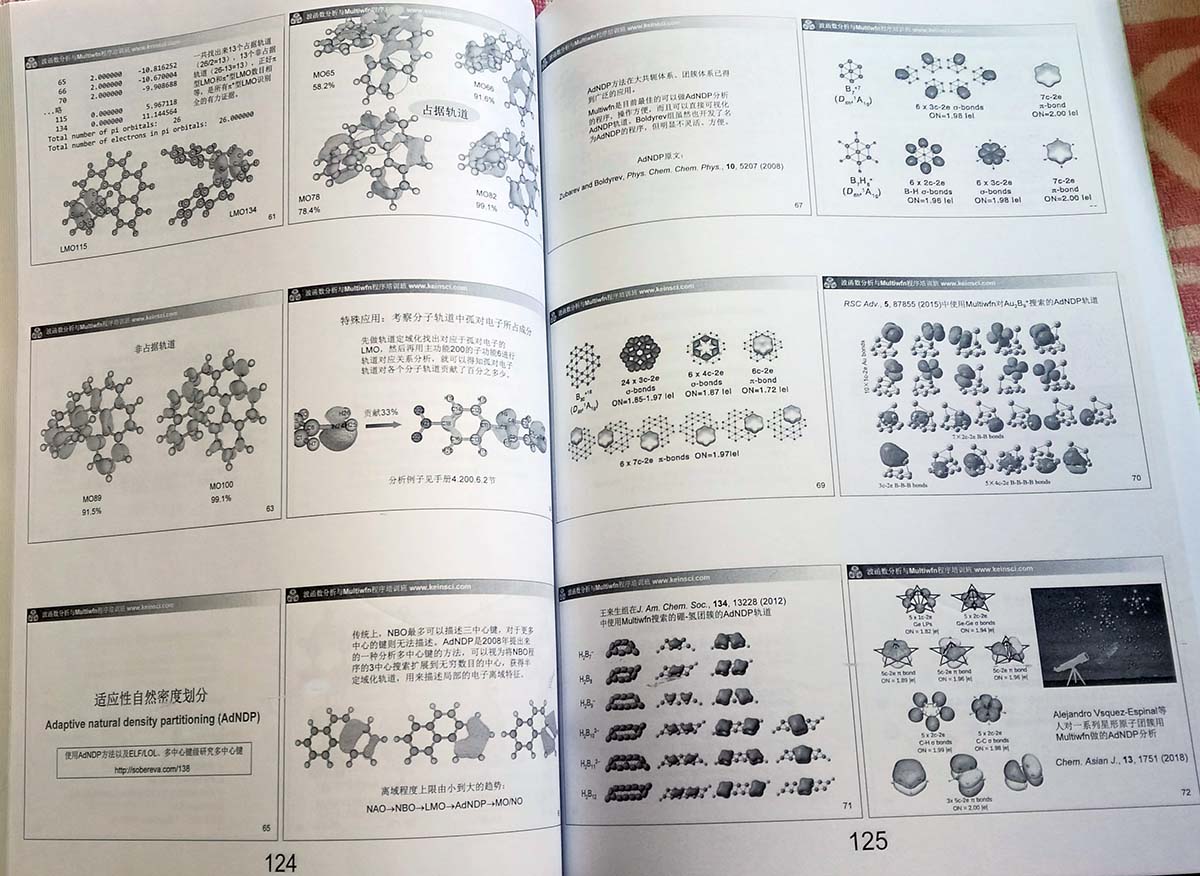
- 轨道定域化、氧化态、AdNDP与BOD/NAdO分析(110多页):介绍定域化分子轨道的概念,以及不同的轨道定域化方法(Boys、Edmiston–Ruedenberg、Pipek–Mezey、NLMO等)的原理和异同。之后通过数个实际体系,演示如何用Multiwfn做轨道定域化分析考察体系电子结构特征。之后介绍如何基于定域化轨道计算任意类型轨道的pi成份。然后介绍氧化态的计算方法,其中重点介绍在Multiwfn中支持的基于定域化轨道的Localized orbital bonding analysis (LOBA)方法,以及卢天对其重要改良的mLOBA方法,通过许多实例展现此方法的重要用处。接下来介绍广为使用的获得半定域化轨道的适应性自然密度划分(AdNDP)方法,会通过多个例子解释如何在Multiwfn中做AdNDP分析考察体系多中心相互作用特征。之后讲解将键级与实空间函数、轨道图景直接相联系的键级密度(BOD)与自然适应性轨道(NAdO)分析方法。
- 电荷分解分析(CDA)(46页):介绍对于考察片段间电荷转移内在细节十分有用的CDA方法、扩展的CDA方法(ECDA)和讲授者提出的广义化的CDA方法(GCDA)。然后介绍轨道相互作用图的用处和价值。之后讲解如何在Multiwfn里做CDA分析以及分析时候的要点,并将通过开壳层、闭壳层、多片段三种类型体系演示具体操作、解读输出数据,并且示例绘制轨道相互作用图的方法以及如何调节作图效果。
- 波函数分析中的实空间函数(约290页):本节信息量巨大,分为好几大部分,与化学键的研究密切相关
(a)实空间函数分布的图形展现方式:介绍实空间函数的概念,以及展现其分布特征的各种方式(曲线图、填色图、等值线图、地形图、梯度线图、向量场图、等值面图、填色等值面图等)
(b)电子密度与自旋密度:详细介绍电子密度、自旋密度以及与之相关的函数的定义和特征,介绍电子密度差的定义和用处
(c)详细介绍电子相关问题的本质,涉及对密度、交换相关密度、相关因子函数、条件概率、相关穴等概念。为之后内容做铺垫
(d)介绍电子的定域性、离域性、离域化指数、定域化指数指数等概念,并结合具体体系令学员直观领会这些概念,然后对定域化分子轨道和电子离域性之间的关系进行辨析
(e)详细介绍电子定域化函数(ELF)的物理意义,并结合大量实际体系展现ELF的特点以及能解释的问题。之后介绍与ELF用处相近的函数,包括定域化轨道指示函数(LOL)、SCI、LED、ELI、EPLF、RoSE、PS-FID、SEDD、DORI、EDR等,其中大多数都被Multiwfn支持。然后介绍ELF-sigma和ELF-pi的定义和实际用处
(f)先介绍范德华表面的定义,然后介绍分子表面的定性和定量分析概念,之后系统性地介绍分子静电势(ESP)的定义、分布特征及其能考察的大量化学上感兴趣的问题
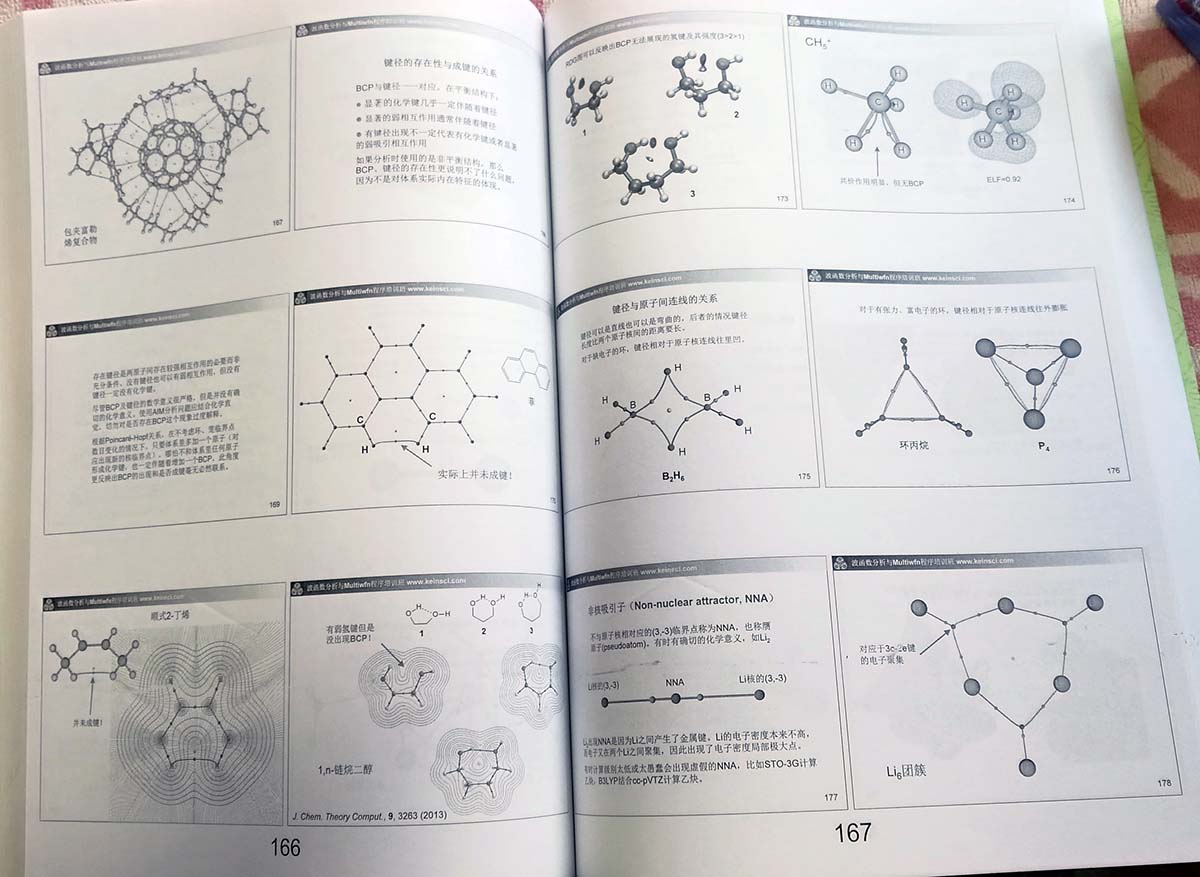
(g)拓扑分析与盆分析:这是实空间函数的高级、定量分析手段,将结合实际分子介绍这类分析中涉及的各种概念,诸如临界点的定义和分类、Poincaré-Hopf关系、键径、非核吸引子、盆、盆间面、吸引子、定域化域、可约与不可约域、二分点等。之后介绍原子的单、偶、多极矩、极化率的概念
(h)其它实空间函数的含义与应用:介绍分子电场、电子密度的拉普拉斯函数、动能密度、势能密度、能量密度等函数,介绍AIM盆的性质,介绍如何通过作图和考察键临界点上的各种实空间函数特征(如键椭率、eta指数、平面性指数、键金属性)讨论问题,比如对相互作用类型进行划分、判断氢键存在性、判断氢键类型、估计氢键键能等。
(i)价层密度分析。这是讲授者提出的考察电子结构的方法,将通过诸多实际体系,展现价层密度分布特征、解读其蕴含的信息,并且与ELF、电子密度拉普拉斯函数、变形密度进行对比来展现异同。
(j)范德华势分析。这是讲授者提出的十分直观、方便、实用的分析范德华作用主导的弱相互作用的方法,是对流行的静电势分析的关键性的补充。将介绍范德华势的概念、在Multiwfn中的实现,并给出诸多例子体现出范德华势分析的极为重要的价值。
(k)介绍Grandinetti等人提出的基于能量密度等值面和键临界点属性判断化学键类型、考察氢键特征与强度的重要方法GLED (Graphic representation of the Local electron Energy Density) - 计算特定点的属性以及绘制一维、二维、三维图像(150多页):这一部分深入详细介绍怎么在Multiwfn里获得三维空间中指定位置处的各种实空间函数的值以及导数信息、怎么计算一条直线上的性质并绘制成曲线、怎么绘制各种类型的平面图考察实空间函数在特定平面上的分布、如何计算某个空间范围的格点数据并绘制等值面图。演示的时候并不仅仅讲程序操作,而是结合各种实际分子和不同的实空间函数,令学员在了解操作的同时体会实空间函数分析的巨大价值以及Multiwfn程序的极强的灵活性。其中还传授许多使用技巧。之后介绍Multiwfn的自定义操作,由此可以轻易地得到诸如密度差图、分子间轨道重叠图等。然后介绍初属性与变形属性的概念以及绘制方法,从而能够考察原子成键过程中电子密度、静电势等函数的变化情况。这部分还演示如何将Multiwfn与VMD联用非常简单快速地绘制效果极佳、对实际研究非常有用的分子表面静电势图。最后介绍如何利用Multiwfn绘制对于研究分子(超)极化率内在特征极其用的(超)极化率密度图。
- 实空间函数的拓扑分析(70页):详细介绍Multiwfn的拓扑分析模块的使用方法,并且给出诸多例子(包括周期性体系),使学员掌握如何使用Multiwfn做电子密度、ELF、ELF-pi等函数的拓扑分析,从而探究化学键和弱相互作用的强度、类型等问题。其中还会讲如何将Multiwfn与VMD结合使用快速来绘制高质量AIM拓扑分析图,如何绘制电子密度等值线+梯度线+临界点+拓扑路径+盆间面的平面图,如何利用约化密度梯度考察电子密度临界点的存在性等问题。之后还会具体讲NBO中的NBCP分析(将NBO的思想与AIM理论中的BCP分析相结合),也同时讲如何在Multiwfn中把临界点属性分解为轨道贡献。
- 实空间函数的盆分析(40多页):将介绍盆分析的算法以及在Multiwfn中的实现。介绍如何使用Multiwfn的高效、普适、灵活、强大的盆分析,结合电子密度、ELF、静电势、密度差、源函数、价层密度等函数以不同角度探究体系的电子结构特征。
- 定量分子表面分析(106页):首先讨论与静电势的定量分子表面分析有关的主题,包括静电势与分子间相互作用朝向的关系、静电势与相互作用能的关系、静电势与结合倾向性的关系、基于静电势的共晶虚拟筛选、基于分子表面静电势定义的描述符、基于这些描述符对物理化学性质进行预测(蒸发焓、升华焓、分子晶体密度、沸点、水合自由能等)、静电势分布面积统计分析。之后介绍平均局部离子化能、局部电子亲合能的概念以及其在分子表面上的分析的重要用处。然后介绍一系列主题,包括讲授者提出的局部定量分子表面分析、定量分子表面分析的数值实现方法以及讲授者提出的简化算法、利用Multiwfn+VMD绘制分子表面静电势的方法等。通过一系列具体实例演示在Multiwfn中对静电势、平均局部离子化能乃至福井函数做定量分子表面分析以获得各种对化学研究重要的信息,还通过示例演示用Multiwfn预测炸药FOX-7的密度的过程。最后专门讨论如何衡量分子的极性。
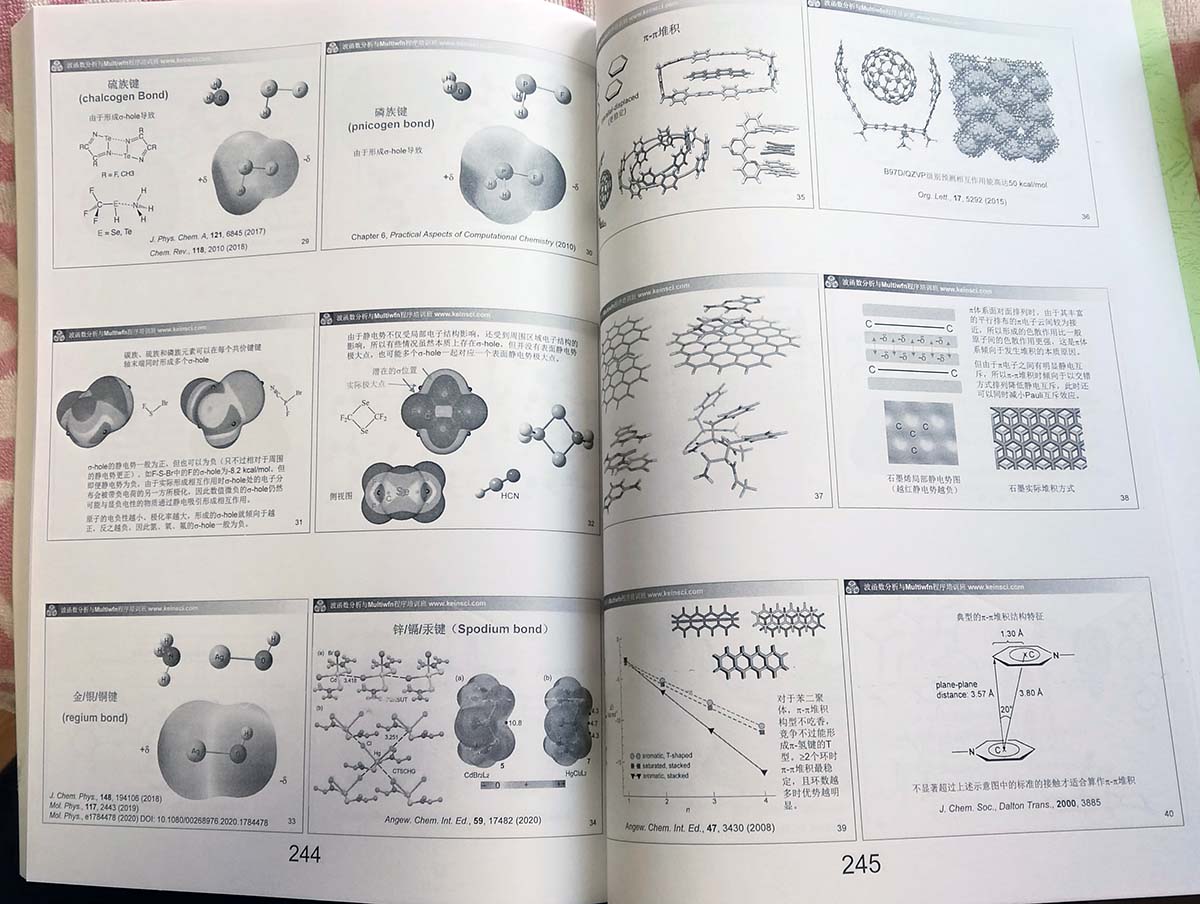
- 弱相互作用分析(207页):首先系统性地介绍氢键、pi-pi堆积、二氢键、卤键、硫键、磷键、阳/阴离子-pi作用等各种弱相互作用类型的特点和本质,然后介绍如何用各种分析方法对弱相互作用的特征、强度进行考察,其中着重讲解和演示约化密度梯度(RDG)、RDG扩展到动态环境下的方法(aRDG)、IRI(卢天提出的相互作用区域指示函数)、DORI、IGM(独立梯度模型)、IGMH(卢天提出的基于Hirshfeld划分的IGM方法)、mIGM(卢天提出的modified IGM方法)、amIGM(卢天提出的将mIGM扩展到动态环境分析的方法)、IBSI和IBSIW指数、Becke/Hirshfeld surface分析、范德华表面穿透特征分析、原子核位置静电势变化分析、分子表面LEAE极小值、CVB指数、LOLIPOP指数等方法的原理和操作,并且结合许多实例对这些分析中涉及的技巧进行介绍。
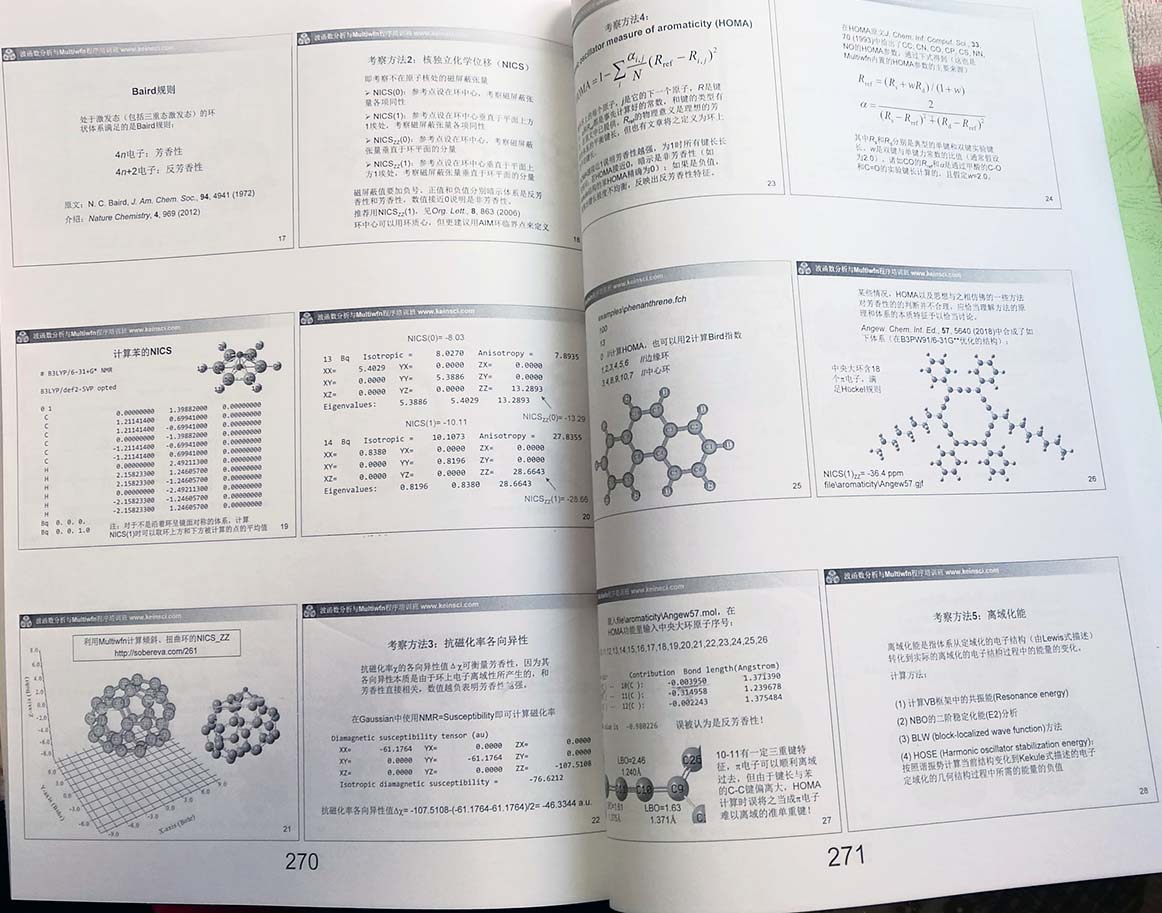
- 芳香性与离域性分析(180页):介绍芳香性的本质、相关概念(如芳香性驱动的反应)、各种衡量芳香性和电子离域性的方法及具体计算操作(含周期性体系),包括休克尔规则、Baird规则、Möbius规则、Craig型芳香性、三维芳香性、同芳香性、双芳香性、核独立化学位移(NICS)及各种变体、NICS的一维扫描和积分、FiPC-NICS、NICS的二维扫描、抗磁化率各向异性、HOMA、HOMAc、HOMER、离域化能(包括介绍使用NBO程序计算垂直共振能的方法NBO-RE)、芳香稳定化能(ASE)的概念以及基于异构化稳定化能获得芳香稳定化能的方法、多中心键级、对位离域化指数(PDI)、ΔDI、芳香性波动指数(FLU)、AIM环临界点处电子密度及其垂直于环的曲率、Shannon芳香性、ELF-sigma和ELF-pi、信息理论定义的芳香性指数、AV1245/AVmin指数及sigma和pi分离、Clar规则、离域电子分布的均匀性与芳香性的关系,等等。之后详细介绍磁感生环电流的概念及其衡量芳香性的方式、相对于NICS的各种优点,然后讲解磁感生电流密度的各向异性(AICD)方法的原理以及实现AICD分析的同名程序的使用,还介绍通过GIMIC和ParaView绘制各种形式的图考察环电流分布特征的方法,简介基于CTOCD方法考察感生电流及相关属性的SYSMOIC程序,说明轨道的感生电流对测量原点的依赖性问题。强调诸多使用磁性质讨论芳香性常被忽视的重要问题。讲解对图形化考察芳香性极为有用的等化学屏蔽表面(ICSS)的原理以及在Multiwfn中的计算方法。通过这一节,学员将对芳香性的概念和分析手段有非常全面充分的认知,从而能够游刃有余地研究芳香性问题。
- 电子激发分析(160多页):介绍和演示Multiwfn支持的各种电子激发分析方法,它们对于描述和探究电子激发的特征和本质有极为重要的用处。内容包括介绍空穴-电子分析(可视化空穴和电子分布、可视化交叠函数和密度差、计算激子结合能、显示分子轨道/原子轨道/原子/片段对电子和空穴的贡献等)、考察跃迁密度的和跃迁偶极矩密度、通过热图讨论跃迁密度矩阵与跃迁偶极矩矩阵、将跃迁偶极矩分解为MO对/基函数/原子的贡献、对密度差进行变换分析电子激发特征、计算Δr和Λ指数、自然跃迁轨道(NTO)分析、计算Ghost-hunter指数判断Ghost态、图形化展现分子片段对跃迁偶极矩的贡献、通过IFCT方法计算任意片段间电荷转移量以及考察电子激发中电荷转移特征百分比、电荷转移光谱(CTS)分析、计算轨道重叠度与质心距离、基于电子激发角度的电子密度极化分析方法、通过LOL函数分析电子激发特征、计算原子Mulliken跃迁电荷及TrESP电荷考察激子耦合、双正交化分析、快速显示一批激发态中的主要轨道跃迁。最后还对Multiwfn的灵活强大的绘制电子光谱以及振动光谱的功能进行介绍。
- 反应位点的预测与反应活性分析(120多页):介绍各种实用的预测反应位点的方法,如福井函数、福井势、简缩福井函数、核福井函数、键反应性描述符、基于NBO的键反应性指数、双描述符、简缩双描述符、双描述符势、轨道权重福井函数/双描述符、(准)简并福井函数/双描述符、键双描述符、分子表面的静电势/平均局部离子化能/局部电子亲和能/局部电子附着能的极值点分析、pz原子轨道布居数分析、前线轨道理论、原子电荷分析、超离域度、FED、Parr函数、FF-EP index(RAI函数)、电子密度对温度的响应函数、HOMOL/LUMOL等。同时也介绍很多概念密度泛函理论的知识,比如电负性、化学势、软度、局部软度、局部超软度、相对软度、硬度、超硬度、亲核指数、局部亲核指数、亲电指数、亲电描述符ε,等等。最后介绍使用Multiwfn非常容易地批量快速计算出所有概念密度泛函理论定义的量的方法。
- 处理格点数据(40多页):介绍Multiwfn十分灵活强大的格点数据处理功能,对于其中极具价值的子功能还给出应用例子,包括屏蔽片段间非交叠区域RDG等值面、格点数据相互运算、对格点数据进行统计分析、计算和绘制(局部)积分曲线和平面平均曲线、考察分子间重叠密度等。
- 批处理与制作动画(26页):介绍Multiwfn的安静模式以及批处理方式运行的方法,然后提供几个例子展现批处理运行的强大和便利,包括绘制RDG填色等值面图的旋转动画表现弱相互作用、绘制乙炔三聚成苯过程的键级变化曲线、制作ELF等值面动画、制作描述变形密度变化的平面图动画。
- 体系结构特征的分析(约60页):介绍Multiwfn支持的表征体系结构特征的各种分析,包括计算各种基本几何参数(质心、回转半径、转动常数、惯性矩等),计算体系的长、宽、高和直径,分子平面性的定量考察和直观展现,计算分子的范德华体积,计算分子或片段的范德华表面积,用域分析展现体系孔洞形状及计算其体积,对晶胞或分子动力学模拟的盒子图形化展现自由区域并计算其体积,计算分子/晶体孔洞直径,计算原子连接关系与原子配位数,计算平均键长和平均配位数,计算共轭链的键长/键级交替指数(BLA/BOA)以及绘制键长/键级随键的序号变化的折线图,动力学直径的计算,分子和固体表面的距离投影图的绘制,分子球形度的计算。
- 能量分解(约100页):介绍各种主流能量分解的基本原理,包括sobEDA/sobEDAw、SAPT、XSAPT、Morokuma、NEDA、LED、ALMO、IQA、Mayer、基于分子力场的能量分解。其中,对Multiwfn开发者提出的特别高效、普适、精确、方便而且特别流行的sobEDA/sobEDAw的原理和思想进行详尽的说明,演示其使用方式,并给出完整计算例子。还详细演示通过Multiwfn做基于力场的能量分解(EDA-FF)并将原子贡献可视化。还介绍原子对色散能的贡献以及色散密度的概念、计算和可视化方法。还对刘述斌教授提出的steric-electrostatic-quantum能量分解方法进行介绍并演示操作。最后,对"相互作用量子原子(IQA)"方法做深入介绍,解读AIMALL程序做IQA分析的输出,给出实际分析例子。
- 通过柔性键力常数考察相互作用强度(26页):讨论衡量键强度的各种指标的共性与差异,之后介绍柔性力常数方法的算法及其讨论键强度方面的重要价值。之后介绍实现柔性力常数分析的compliance程序的安装、使用,并给出一系列分析实例。最后简要介绍另一个局部力常数分析程序LModeA-nano。
- 其它NBO分析方法(70页):详细介绍NBO中的一些高级分析方法的概念和具体使用,包括deletion分析、自然键-键极化率(NBBP)、自然位阻分析(Steric分析)、自然簇分析(NCU)、自然库仑静电分析(NCE)、自然化学位移分析(NCS,包括介绍基于NCS方法对NICS值进行分解的方法)、自然J耦合分析(NJC)、输出系数矩阵/算符矩阵的方法和MATRIX关键词的使用、偶极矩分解分析(DIPOLE分析)、其它单电子属性分解分析,以及其余一些NBO中有价值的关键词的使用。
- 使用Multiwfn绘制光谱图(50多页):介绍和演示用Multiwfn强大的光谱绘制模块绘制各种类型的光谱(红外、Raman、VCD、ROA、UV-Vis、ECD、NMR),强调Multiwfn在光谱绘制方面的独特长处,包括构象权重光谱的绘制、多个光谱的同时绘制、将总光谱拆分成不同跃迁的贡献、用竖线体现不同跃迁对应的位置、自动标注光谱峰位置、自动大批量绘制光谱、基于UV-Vis光谱预测准确物质的颜色等等。
- 修改Multiwfn的代码(50多页):讲解Multiwfn的编译方法,详细介绍Multiwfn程序的各个源文件、程序整体架构、程序中重要的变量/数组的含义、波函数信息的记录规则、重要函数的用处,并给出修改程序源代码实现特殊功能的实例。Multiwfn代码简单易读、结构清晰,再加上本部分的学习,学员就可以对Multiwfn代码自如地进行修改和扩展。
- 充分运用波函数分析的理论计算文章的模板(约50页):这部分介绍如何利用量子化学计算和丰富的波函数分析,结合恰当的选材,写一篇题名为诸如A theoretical investigation on XXX: A DFT的内容充实又有深度的研究文章。这部分绝非让学员灌水(灌水可耻!),而是以文章模板的形式,令学员懂得面对不同的问题都适合做什么样的理论计算和波函数分析,使其能够结合实际研究回忆和综合运用所学知识、加深印象和理解,充分达到学以致用的目的!
为了让大家更好地了解本培训班的超高含金量,这里把培训讲义中随意拍的15页内容贴出供大家参考。本培训内容是波函数分析领域的知识、讲授者的研究经验和成果的集大成之作。