第2届北京科音高级量子化学培训班获得圆满成功!
2025年10月16日
北京科音自然科学研究中心(http://www.keinsci.com)于2025年10月11日至14日在北京国润商务酒店会议中心成功举办了“第2届北京科音高级量子化学培训班”(报名页面见http://www.keinsci.com/workshop/2KAQC),本培训讲授者为北京科音自然科学研究中心主任卢天(http://www.keinsci.com/members/Tian_Lu.shtml)。本次培训已经是北京科音从2015年以来举办的各类计算化学培训中的第67届。北京科音的培训以其非常丰富和充实的内容、极高的含金量和实用性、认真的态度以及良心的收费而著称,深受历届参加者的好评,也因此在中国的计算化学领域获得了非常好的口碑和很高的关注度,访问http://bbs.keinsci.com/forum-43-1.html可阅览往届所有培训的回顾和学员评论。本次培训4天的课程同往届一样在一片掌声中顺利、圆满结束!
参加本次培训的学员共170多人,其中约100人到现场参加,其他的学员通过快递寄送讲义+听全程现场录音+培训专用群里答疑方式学习,也有不错的学习效果。
本次培训的学员来自全国各地高校,如:清华、北大、兰大、南大、上交大、西北大学、天大、南开、大连理工、浙大、中科大、吉大、暨南、川大、集美、人大、北师大、北海艺术设计学院、哈工大、北科大、北理工、同济、山西大学、皖西学院、北化工、武大、山大、南科大、中国人民解放军海军军医大学、国科大,等等。
学员也有很多来自研究院所,如:中国科学院过程工程所/赣江创新研究院/福建物构所、国防科技创新研究院、中国原子能科学研究院、北京科学智能研究院、中国中医科学院中药研究所、国家纳米科学中心、内蒙合成化工研究所,等等。
还有不少大陆以外学校的学员参加,包括:乌特勒支大学、澳门大学、Texas A&M、马来西亚大学、曼彻斯特大学、纽约州立大学布法罗分校、北达科他大学、香港中文大学、杨百翰大学。
也有少量学员来自与化学、药物、生物、能源、材料有关的单位:InnoLux (群創光電)、绿能起源科技、微共聚新材料、南充市聚邦科技、中国石油天然气股份有限公司、北京望石智慧、广东迪美生物、江西钨业集团。
本培训的目的是使得已经具有北京科音中级量子化学培训班(http://www.keinsci.com/KBQC)或相似水平的有经验的研究者显著进一步提升研究能力,在短时间内能从“跑”的水平真正达到“跳”的水平,提供一个从老鸟进阶为高手的最佳的直升梯!通过本培训,学员们掌握了远比中级班涉及的更高级、更深入、更复杂的理论方法和研究问题的手段、学会了更多问题的研究方法,如CASSCF和多参考计算、从头算动力学、全电子相对论计算、振动电子光谱的计算、势能面交叉与光化学涉及的MECP和MECI搜索、磷光速率的计算、振动分辨的吸收/荧光/磷光/ECD/CPL光谱的计算、内转换与系间窜越速率的计算、低标度耦合簇方法、显式相关计算、sTDA极快速大体系光谱计算、(MR)SF-TDDFT方法的原理与应用、DFT泛函的调控、real-time TDDFT模拟、VSCF/VCI等高级振动分析方法、双光子吸收的计算、超高精度热力学量计算需要考虑的DBOC项的获得,等等。学员还同时学会了ORCA、xtb、sTDA、Dalton、GAMESS-US、MRCC、NWChem、eT等更多计算程序的使用。通过本培训学员们无疑能在量子化学研究方面更上N层楼,研究更广泛的问题,做出明显更有深度、更出彩的研究工作!本培训里讲的内容都是讲授者卢天老师长年累月花费海量时间研究和总结出的极难以其它方式获得的珍稀干货,十分硬核,信息量浩瀚!本培训完整讲授内容见本文末尾的附录。
本次课程的安排超级紧凑,信息密度和授课强度超级大,卢老师在课上每天上午3小时、下午5个多小时不留余力地倾囊传授知识和经验,连一次课间休息都没有,每天结束时间还比预计时间拖堂了约半小时以尽全力传授更多知识。本培训还提供了大量电子资料(综述、重要论文等)以供对不同主题感兴趣的学员进一步深入学习。为了尽最大可能讲好信息量超大的本培训内容,培训期间卢老师每天都全力备课到深夜,甚至将近凌晨五点。
在本次现场培训结束后,每天卢天老师都在培训QQ群里花大量时间及时、认真、耐心地回答学员们提出的各种听课时产生的问题以及自己平时研究中遇到的困惑。在限时的培训专用QQ群解散后,学员们的问题还将会继续在规模巨大、特别专业的综合性计算化学交流论坛“计算化学公社论坛”(http://bbs.keinsci.com)和计算化学领域十分知名、活跃度极高的超过一万人的QQ群“思想家公社”(http://sobereva.com/QQrule.html)中得到卢老师无限期的解答。参加包括本培训在内的北京科音举办的各种计算化学培训,可以直接申请成为思想家公社QQ群的VIP成员,卢老师在群里解答他们的问题时的优先级和详细程度远高于普通群成员!
值得一提的是,为了令学员们在本次培训中能获得比2024年10月举办的第1届高级班时更多的知识,本届培训前卢老师耗费了两个半月呕心沥血制作了巨量新幻灯片,并大幅更新和完善了原有内容。本次的幻灯片达到了超过2100页!相关输入输出文件超过2600个!信息量比第一届多了近50%!!!培训时间也从3天扩展到了4天,而本届的学生自费价格(1750元)却仅仅比第1届提升了不到10%,费用实在良心。同样的培训内容如果换成利益熏心的商业机构举办(时下多得很),至少要拆成5个培训,总收费至少达到两万,比如本培训中的CASSCF和多参考计算部分单独弄一个培训、从头算动力学和real time TDDFT部分单独弄一个培训、势能面交叉与光化学+内转换和系间窜越单独弄一个培训、ORCA的使用弄一个培训... 本培训的含金量和收费,与如今一些机构甚至就只讲一些量子化学计算皮毛的水培训也要收费三、四千元的行为形成了极为鲜明的对比。对于那种追求利益最大化、没有传播知识造福学员理念、不在乎在内行群体中口碑的纯商业机构的做法,北京科音是极度不屑、十分看不起的。参加本次高级量子化学培训班的学员都是真真正正懂量子化学且非常有辨识能力的专业的研究者,能一次性收获如此极难在其它地方学到的海量宝贵知识、干货,实在是太幸运!
未来的培训:北京科音自然科学研究中心每年都举办不同层次的量子化学培训,以传播量子化学知识、提升我国量子化学研究者的整体水平。本届培训的讲义、录音、相关电子文件现已可以联系北京科音官方购买,以供没赶上本次培训的研究者自学,详见http://www.keinsci.com/KAQC中的说明。下一届(第3届)高级量子化学培训班预计于2027年。欢迎关注北京科音官网(http://www.keinsci.com)首页的预告栏,并且请关注“北京科音”微信公众号,未来培训的预告都会在上面通知。北京科音举办的各种培训相关问题请参见《北京科音办的培训班FAQ》(http://www.keinsci.com/FAQ.html)。
接下来北京科音即将举办的培训是“第17届北京科音分子动力学与GROMACS培训班”,将于11月1日至4日于北京举办。这是真正一次性全面、系统地掌握分子动力学模拟方法和GROMACS分子动力学程序使用的不可错过的培训,既适合从头入门,也同样很适合查缺补漏进一步提高,报名正在进行中,欢迎访问http://www.keinsci.com/workshop/17KGMX查看详情和参加!
以下是本次培训的现场照片













培训令学员们获益匪浅,在培训结束后有很多学员都找卢老师在讲义上签名

卢老师(Sobereva)无论是在培训内容还是课程内容答疑上,都追求最高质量。课后在培训专用QQ群里卢老师及时、认真回答每一位学员的问题,例如:
以下是本次第2届北京科音高级量子化学培训班内容的完整介绍,幻灯片页数一起给出:
- 密度拟合/RI与COSX技术(20多页):这部分讲解ORCA等程序中对加速DFT、后HF、TDDFT等计算起到至关重要作用的密度拟合/RI与COSX技术,只有懂得了这些知识才能将这些程序真正用得又好又快。内容包括:先回顾交换项和库伦项的计算公式,然后讲解密度拟合/RI近似的思想和实现细节、RI技术用于MP2/双杂化和后HF、辅助基组(/J、/JK、/C型辅助基组的意义和选择)、split-RI-J、Cholesky decomposition技术、chain of spheres exchange (COSX)技术的原理和实际价值、各种量子化学程序对加速技术的支持情况。还讲解被很多程序用于加速库仑作用计算的Fast multipole moment (FMM)技术,以及ORCA 6.1开始引入的加速巨大体系库伦计算的Bubblepole (BUPO)技术
- ORCA量子化学程序的使用(325页):ORCA是达到量子化学高手阶段必须充分掌握的程序,这一节真是把ORCA讲得极尽全面和透彻,通过这部分内容学员可以非常快速、顺利上手强大、高效、免费、日趋流行的ORCA程序的主要功能的使用,并获得大量非常重要的卢老师总结的使用经验,从而达到用得游刃有余的程度。通过本培训学远比自己啃手册、搜集网上零七八碎资料学习ORCA顺利太多了!本部分内容如下
• ORCA的基本特征:ORCA的历史与现状;盘点ORCA的优、缺点;罗列ORCA的相关网络资源;盘点ORCA的主要功能和各种其它功能;盘点ORCA支持的所有理论方法;ORCA对核坐标的解析导数的支持情况;Gaussian用户使用ORCA的几个需要注意的问题;罗列ORCA的各种子程序以及附带的独立运行的程序的功能;ORCA的并行与计算资源的设定;ORCA的安装与运行;批量运行ORCA;介绍gbw和其它常见的临时文件
• ORCA的输入文件:使用Multiwfn创建输入文件;输入文件的基本规则;内坐标的输入;理论方法的输入;色散校正的设置;冻核设置;Grimme提出的重要的各种3c系列方法介绍(如B97-3c、r2SCAN-3c、ωB97X-3c等等);内置基组的输入;自定义基组与赝势的输入;RI和COSX的利用;辅助基组的指定;常用关键词;SCF收敛限;SCF的模式;积分格点设置;对称性的利用和对优化的影响;多步任务;使用隐式溶剂模型,包括CPCM、SMD、openCOSMO-RS、DRACO
• 单点计算与相关问题:普通泛函ωB97M-V结合def2-QZVP计算示例(包括输出信息详细解读、观看轨道、依靠Multiwfn做布居和键级分析);双杂化泛函PWPB95-D4计算示例;精度非常高的ωB97M(2)双杂化泛函的特殊使用方法;单参考体系的金标准CCSD(T)计算示例;MDCI模块给出的耦合簇的密度种类的说明;AUTOCI模块支持的高级别耦合簇CCSDT计算示例;圣杯方法Full CI计算示例;轨道优化的微扰和耦合簇计算示例;深度全面盘点ORCA遇到SCF不收敛、收敛异常的解决方法(约20种!)
• 几何优化;收敛标准和自定义;B97-3c优化弱相互作用复合物的例子(输出文件详细解读、输出的各种文件盘点、收敛情况的可视化监控,其中利用到卢老师开发的OfakeG程序http://sobereva.com/498);用ORCA支持的CCSD(T)解析梯度做优化;限制性优化的方法,其中还包括令优化时特定片段以及每个分子都保持刚性的方法;几何优化不收敛、难收敛的解决方法盘点(10种)
• 振动分析:振动分析的功能;orca_vib的用法;振动分析任务输出文件解读;演示ORCA结合卢老师开发的非常流行的Shermo程序(http://sobereva.com/552)算热力学数据;观看振动模式动画;用ORCA+Multiwfn绘制红外、Raman和振动圆二色谱(VCD);盘点约10种虚频的解决方法
• 势能面扫描:做刚性扫描的方法和RI-MP2下做扫描的具体例子;Morse势函数的自动拟合;做柔性扫描的方法和r2SCAN-3c做键拉伸扫描的具体例子
• 优化过渡态:介绍optTS、scanTS和NEB-TS三种找过渡态的方法;以HF加成到乙烯为例演示optTS找过渡态
• 产生IRC:介绍跑IRC的方法、给出具体实例、对结果分析绘制
• 用NEB-TS同时产生过渡态和反应路径:以oxycope重排为例,非常完整地示例怎么用ORCA特有的NEB-TS方法基于反应物和产物结构在产生反应路径的同时得到过渡态,并对细节进行说明
• 电子激发与电子光谱计算:TDDFT计算的重要选项盘点;关于积分的考虑;溶剂效应的考虑;TD-PBE0/def2-SV(P)结合CPCM描述溶剂环境计算螺烯的激发态的例子;TDDFT对苯胺S1激发态做优化和振动分析的例子;TDDFT计算激发态偶极矩的方法;ORCA+Multiwfn绘制UV-Vis谱、电子圆二色谱(ECD);双杂化泛函做TDDFT的方法,以及TD-DSD-PBEP86/def2-TZVP计算卟啉的激发态;ORCA特色的DFT/ROCIS方法(对开壳层体系可以得到自旋纯态激发态)的特点、用法,以及计算苯胺自由基的无自旋污染的二重态和四重态激发态的例子;高精度EOM-CCSD方法的使用和计算例子;比EOM-CCSD标度更低而且算电荷转移激发更好的STEOM-CCSD的使用和计算例子;ADC(2)算激发态的用法;ORCA结合Multiwfn预测物质的颜色;通过MOM方法做ΔSCF方式的激发态计算的原理和实例
• 属性计算:示例CAM-B3LYP/def2-TZVPPD计算18碳环的静态极化率、RI-MP2/aug-cc-pVTZ计算水的静态极化率、CCSD(T)解析计算水的静态极化率;DFT计算动态极化率;DFT计算第一超极化率;NMR的计算方法,以及用计算NMR很好的双杂化泛函revDSD-PBEP86结合pcSseg-2基组的计算例子,以及结合Multiwfn绘制NMR谱;核自旋-自旋耦合常数的计算例子;NICS的计算;能计算深层电离能的IP-EOM-CCSD的使用和例子;能计算各阶电子亲和能的EA-EOM-CCSD的使用和例子
• 杂知识和杂计算:利用libXC使用比ORCA内置的更多泛函的方法以及实际例子;ORCA中双电子积分的储存;ORCA的json文件的意义和产生方法,并示例将AUTOCI模块产生的CCSD(T)级别的单电子密度矩阵写入json文件;“免费”的BSSE校正方法gCP的使用;counterpoise校正在ORCA中的实现;ORCA的便利的自动基组外推的详细介绍和使用技巧;利用ORCA的compound script做热力学组合方法计算;分数占据数的DFT计算;有限温度的DFT计算;快速、直观展现不同区域静态相关强弱的FOD分析的原理,以及在ORCA中的实际计算例子和使用Multiwfn可视化FOD的例子;加外电场;加背景电荷;加外力;初猜轨道的交换;对称破缺单重态的计算(guessmix、FlipSpin和BrokenSym方式),以及反铁磁性耦合常数的计算;波函数稳定性测试和自动优化稳定波函数;产生MP2、CCSD、CASSCF、MRCI自然轨道以及结合Multiwfn产生TDDFT自然轨道的方法;产生UNO轨道的方法;输出Fock矩阵、密度矩阵、重叠矩阵等各种矩阵的方法;orca_plot程序简介;ORCA与Gaussian的联用;极小能量圆锥相交(MECI)搜索功能说明;简介卢老师开发的非常流行的团簇构型和分子构象搜索程序molclus(http://www.keinsci.com/research/molclus.html)与ORCA的结合;使用ORCA做多尺度计算的介绍;ORCA做ONIOM(ωB97X-3c:GFN2-xTB)搜索姜黄素的H转移过渡态的实例
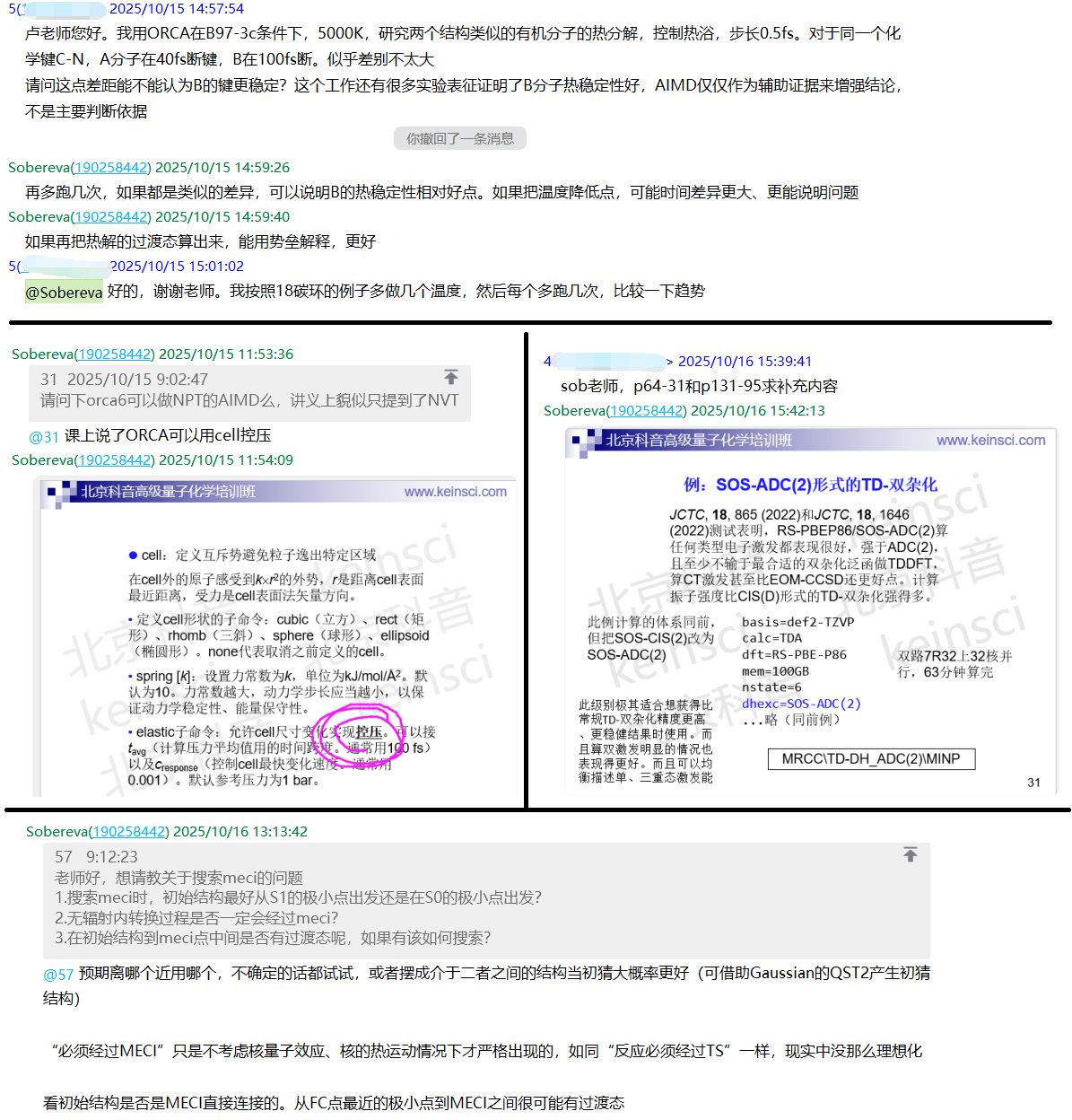
- 使用MRCC和eT做基态和激发态的高精度耦合簇计算(56页):耦合簇是如今使用极其广泛的高精度计算方法,虽然Gaussian等大宗向量子化学程序就能做耦合簇计算,但有明显不足,比如无法做CCSD(T)以上级别的耦合簇计算、不支持超高精度的算激发态的LR-CC3方法、不支持精度好又稳健且性价比比EOM-CCSD明显更高的LR-CC2激发态计算方法,等等。这一节讲授两个以做耦合簇计算为专长的程序MRCC和eT的使用,能完全解决以上问题。还简要提及使用Dalton通过线性响应耦合簇方法高精度计算激发态。本节内容:
• MRCC部分:盘点MRCC的功能和支持的各种方法;MRCC的编译;MRCC的运行;输入文件MINP的写法和重要选项;超高精度的CCSDT(Q)计算能量实例;CCSDT解析梯度优化分子实例;产生精度极高的CCSDT级别的密度以及令其被非常强大的波函数分析程序Multiwfn读取的方法;LR-CCSDT计算极高精度激发能和振子强度实例;高效且稳健的SCS-CC2计算激发能的实例。还顺带介绍MRCC做ADC(2)计算、SOS-CIS(D)形式的TD-双杂化计算,以及MRCC独家支持的SOS-ADC(2)形式的TD-双杂化用法说明和实际计算例子。其中示例的RS-PBEP86/SOS-ADC(2)方法算任何类型电子激发都表现得很好,很适合想获得比TDDFT明显更高精度、更稳健结果且希望计算不过于昂贵时使用
• eT部分:eT是近年来新开发的特别擅长做耦合簇计算的免费、高效的程序,介绍文章见J. Chem. Phys., 152, 184103 (2020)。这部分介绍eT程序的功能、安装、使用方法,重点演示通过eT使用比TDDFT稳健精确得多的Cholesky分解加速的LR-CC2方法以及使用极高精度的EOM-CC3方法计算分子的激发能和振子强度。还演示通过不对称Lanczos链方法结合EOM-CCSD精确高效计算UV-Vis全范围光谱的做法。顺带一提,激发态的CC3计算用eT是最佳选择,远比这方面并行效率糟糕的Dalton快得多。在免费程序里eT是做LR-CC2的最佳选择,MRCC虽然也能做但得不到振子强度 - 低标度耦合簇方法(45页):耦合簇方法可以达到很高的精度,但是基于正则轨道的耦合簇的标度巨高,例如CCSD(T)的标度高达O(N7),导致30原子以上的有机体系都基本算不动。为了能让耦合簇算大体系(大几十原子乃至上百原子),必须使用本节讲的低标度耦合簇方法!本节讲的内容是如今做高精度计算的人必会的。本节重点讲授已经非常流行的高精度计算中等甚至较大体系的DLPNO-CCSD(T)方法,包括:DLPNO方法的概况和发展(包含与MP2、F12等方法或技术的结合)、计算耗时/标度/资源消耗情况、影响精度的关键参数PNO阈值以及LoosePNO/NormalPNO/TightPNO的选择、耗时比tightPNO增加不多但能得到明显更好精度的PNO外推、DLPNO-CCSD(T1)的特点、DLPNO-CCSD(T)计算弱相互作用需要注意的问题和局限性、其它低标度耦合簇方法(MRCC的LNO-CCSD(T)、Molpro的PNO-LCCSD(T)以及它们与DLPNO-CCSD(T)的对比)。详细介绍用ORCA做DLPNO-CCSD(T)的方法,给出DLPNO-CCSD(T)/def2-TZVPP计算瑞德西韦分子的例子、DLPNO-CCSD(T1)/cc-pVTZ计算18碳环的例子、对C8F8分子做DLPNO-CCSD(T)的CPS外推的例子、做DLPNO-双杂化泛函计算的例子(ωPr2SCAN50-D4计算九并苯共价二聚体)。介绍对不小体系高精度计算激发态的DLPNO-STEOM-CCSD方法和在ORCA中的计算实例。给出用MRCC程序做LNO-CCSD(T)计算的方法和例子。最后顺带介绍能够令MP2、耦合簇实现线性标度的cluster-in-molecule (CIM)方法的具体介绍,并给出在ORCA中做CIM-DLPNO-CCSD(T)计算的实际例子
- 显式相关计算(34页):对于经常用后HF方法做高精度量子化学计算的研究者,这一节详细讲授的显式相关计算的知识是必须要掌握的。利用这种技术,使用相对较低的代价就能得到≥ 4-zeta档次昂贵基组结果,因此遇到QZ、5Z档次基组算不动的时候,就特别应该考虑做显式相关计算,其中F12是最流行的形式,早已广泛流行。本节的内容包括:电子相关能随基组尺寸的收敛的介绍;给波函数引入电子间距离项的意义;氦原子的显式相关求解;R12方法;显式相关计算电子积分时对RI的重要利用;F12方法;显式相关方法的性能;显式相关计算用的基组(Dunning相关一致性基组的F12版,误差对比,补偿辅助轨道的概念,补偿辅助基组CABS的意义);显式相关的实际应用;MP2-F12的不同形式;CCSD-F12的不同形式;对耦合簇的(T)项的F12修正;色散权重形式(DW-MP2、DW-CCSD(T**)-F12);CABS singles校正;显式相关计算的BSSE问题;支持显式相关计算的程序盘点;ORCA做F12计算的关键词写法和基组;RI-MP2-F12/cc-pVDZ-F12计算实例;CCSD(T)-F12/cc-pVDZ-F12计算实例;不同显式相关计算方式的结果和耗时对比
- 相对论量子化学计算(123页):量子化学计算牵扯到较重原子时总要考虑相对论效应,不恰当考虑相对论效应轻则精度差,重则结果毫无意义。这一节全面、完整、深入讲解了量子化学中与相对论有关的一切背景知识,结合实例充分讲授了如何在量子化学计算中合理且有效率地考虑相对论效应。注意,通过相对论赝势的方式体现标量相对论效应的做法在北京科音中级量子化学培训班(http://www.keinsci.com/KBQC)中的“赝势与相对论量子化学初步”一节已经十分详细讲过了,本节主要讲的是做全电子相对论计算的方法,虽然更贵,但克服了只靠赝势体现标量相对论效应的严重局限性。本节的内容:
• 相对论量子化学的基础知识:回顾狭义相对论的基本原理和洛伦兹变换;原子序数与相对论效应大小的关系;相对论效应对原子壳层和对化学问题的各种影响;旋轨耦合对价轨道的影响;Dirac方程与spinor(旋量)波函数;分子体系的Dirac方程的哈密顿;四分量相对论→二分量相对论→标量相对论&旋轨耦合→赝势&旋轨势的关系梳理;四分量的量子化学计算方法;四分量变换为二分量处理、Foldy-Wouthuysen (FW)变换、Breit-Pauli哈密顿、质量速度项、单/双电子Darwin项、单/双电子旋轨耦合项;孤立原子体系的旋轨耦合情况;盘点各种二分量哈密顿(DKH、ZORA、FORA、IORA、SEAX、Wood-Boring、RESC、X2C、DLU-X2C、NESC、IOTC);picture change;标量相对论计算;实际研究中旋轨耦合的考虑(考虑单个态和多个态的旋轨耦合效应的场合、意义、做法、实现程序);旋轨耦合算符的近似考虑(Zeff、SOMF、AMFI以及主流程序的支持情况);点电荷和有限核模型;四分量和二分量计算程序盘点
• 相对论计算用的基组:先讲全电子相对论计算的优缺点,然后盘点各种全电子相对论计算可以用的基组,如(aug)-cc-p(wC)VnZ-DK系列、cc-p(wC)VnZ-DK3/-X2C、seg-cc-p(wC)VnZ-IOTC、SARC、SARC2、def2系列重收缩版、x2c-系列、ANO-RCC、ANO-R、Dyall、Sapporo系列、Hirao的DKH3基组、RPF-4Z、Jorge基组、Wahlgren/Faegri (RAF-R)、Cologne DKH2等。并且给出全电子相对论计算使用基组推荐。还提及WTBS、DGauss系列、UGBS、SVPall/TZVPall/TZVPPall这些未对相对论计算优化的全周期表全电子基组。之后讲赝势+旋轨势计算用的赝势基组,包括Stuttgart系列、dhf系列、CRENB系列
• 在ORCA的计算中考虑相对论效应:ORCA对单态和多态的相对论效应的考虑;ORCA支持的旋轨耦合算符;基于X2C在PBE0/x2c-TZVPall结合X2C/J下计算顺铂的例子;基于DKH2在CCSD(T)/cc-pVQZ-DK下计算硅炔的例子;基于DKH2用ωB97X-D4结合SARC-DKH-TZVP和重收缩版的def2-TZVP计算三乙酸铽的例子;基于X2C的全电子标量相对论对Hg2做解析梯度优化和振动分析的例子;基于X2C计算TMS的NMR的例子;旋轨耦合TDDFT计算硫代胸腺嘧啶的例子;旋轨耦合TDDFT计算Os配合物的例子,其中还结合Multiwfn绘制考虑旋轨耦合前后的电子光谱进行对比;CASSCF计算中考虑旋轨耦合考察F原子基态的能级分裂;MRCI计算中考虑旋轨耦合得到OH自由基基态的旋轨耦合分裂能
• Gaussian中做相对论量子化学计算:做全电子标量相对论计算的方法;做全电子二分量相对论计算的方法;做赝势+旋轨势的二分量计算的方法;DKH2标量相对论计算Cu(CO)+的例子;DKHSO二分量全电子计算PbI2的例子;充分考虑相对论效应的高精度焓的计算方式;CASSCF下CH2的T0-S1间旋轨耦合的计算;CASSCF下OH的简并基态间的旋轨耦合的计算;在NWChem中做考虑旋轨势的DFT计算
- 约束性DFT(35页):普通的DFT计算在SCF过程中通常都会收敛到能量最低的解,然而出于特殊目的,研究者往往希望得到具有特定电子结构特征的解,例如要求特定原子或片段上具有特定数目的净电荷或单电子,这必须通过本节讲授的约束性DFT(CDFT)方法实现。本节介绍CDFT的各种相关知识并给出通过NWChem程序做这种计算的丰富的例子。内容包括:CDFT的基本概念;CDFT的实际用处;支持CDFT的程序;CDFT的实现原理和算法细节;NWChem的CDFT功能;CDFT任务输入文件的基本格式;约束的设置方法;CDFT难收敛或报错的解决;检验收敛的波函数的做法。实例1:计算菌绿素-锌菌绿素的电荷转移态(输入文件写法要点、输出文件解读、不同CT态结果的差异、和TDDFT结果的对比、利用Multiwfn程序做布居分析和密度差图检验和理解得到的波函数特征)。实例2:考察苯...Li+的电荷分布与能量的关系的实例,通过shell脚本对电荷进行扫描以研究Li的电荷变化如何影响能量。实例3:巧妙地利用CDFT将闭壳层的二氧杂环丁烷二酮优化成草酸双自由基。实例4:利用CDFT计算吡啶阳离子的正电荷完全定域在N上的状态和真实状态的能量差
- DFT泛函的调控(50多页):这一节着重讲授针对实际研究的体系优化范围分离DFT泛函的关键参数ω的方法,从而令泛函对各种类型激发能、(超)极化率等问题都能有很好的计算结果,这个方法在文献中已使用得十分普遍。内容:回顾纯泛函和低HF成份泛函的普遍问题;交换泛函的长程校正(LC);范围分离泛函的概念;ω的最优调控(IP-tuning);详细介绍ω调控的泛函计算IP、EA、激发能、(超)极化率、光学旋转方面的表现;ω调控的主要缺点;量子化学程序中ω的设置;实现ω调控的程序盘点;使用卢老师开发的optDFTw(http://sobereva.com/346)程序调控ω的方法和实例;使用scanDFTw扫描ω的方法和例子;体系能量与电子数的关系(和离域性/定域性误差密切相关),以及使用shell脚本和NWChem程序绘制能量与电子数关系图的实例;ω+α二维调控;全局密度依赖(GDD)的调控;基于ELF的快速的ω调控方法(ELF调控)以及利用Multiwfn做这种调控的实例;机器学习预测最优的ω;三重态调控(Triplet Tuning);溶剂环境下的ω调控、ω调控的屏蔽的范围分离泛函(SRSH)的效果和实现方法
- CASSCF、多参考方法的原理与应用(360多页):CASSCF和多参考方法是计算考虑静态相关显著的体系基本上离不开的方法,多参考计算说是各种理论方法中的明珠也不为过,而它们又是量子化学计算方法中使用起来最难的,尤其是对结果有至关重要影响的活性空间的合理选取的门道很深。本节对CASSCF和多参考方法相关背景知识讲得极为系统和详细,精心编排的充分涵盖各种情况的计算例子已达到了30多个,从最基础的CASSCF(2,2)到高精度的MRCI/MRCC再到最新的NEVPT4(SD)和ICE-NEVPT2全覆盖、从单原子到有机分子再到过渡金属配合物各种体系全覆盖、从化学反应能/势垒计算到激发能计算到振动分析再到NMR计算等等各种问题全覆盖、MO/NO/NBO/LMO/UNO/有限温度MO/AVAS等各种初猜轨道的利用全覆盖...各个例子都特意强调对相应情况如何恰当定义活性空间和产生初猜轨道。目前这一节的含金量和内容丰富程度说是CASSCF和多参考计算的葵花宝典也不算太夸张,着实是这类方法计算从初识到精通的近乎完美的学习途径!具体内容:
• MCSCF的相关知识:不同方法对动态相关和静态相关的回顾(注:静态和动态相关的概念在北京科音中级量子化学培训班里已经详细讲过了,因此这里不再花时间详谈);MCSCF的原理;CASSCF的原理、活性空间的定义、轨道的旋转不变性;RASSCF的原理、用处以及相关方法GASSCF;MCSCF算多个态需通常要用的态平均的概念,以及“动态”权重方法;全面详细讲解活性空间的选择,分各种情况讲,例如电子激发计算、化学过程计算、单个结构的计算(谨慎判断&粗暴判断&通过自然轨道或有限温度DFT计算的轨道占据数判断)、基于定域化轨道选择、开壳层单重态体系、原子轨道角度的考虑、double-shell效应、辅助确定初始轨道/活性空间的方法(AVAS、GVB、autoCAS等);MCSCF的收敛问题;MCSCF的轨道优化方法;做MCSCF的程序全面盘点
• 多参考方法的相关知识:多参考方法的概念;多参考组态相互作用(MRCI);多参考微扰类方法的全面介绍,包括CASPT2(原理的具体说明、入侵态问题、CASPT2的虚能级移动、CASPT2的IPEA移动、CASPT2-K)、NEVPT2、多参考微扰方法的多态版本(MS-CASPT2、MCQDPT2、QD-NEVPT2、XMCQDPT2、XMS-CASPT2)、多参考四阶微扰方法NEVPT4(SD);多参考耦合簇(MRCC)和MR-EOM-CC;不同方法优化的系数总结;电子相关计算中的轨道优化;其它多参考方法(DCD-CAS(2)、MR-CEPA(0)、CIPT2、SORCI、MC-PDFT、short-range DFT (srDFT)、DFT/MRCI);OO-CCD(T)用于静态相关不强不弱体系的价值的讨论;做MCSCF、多参考计算的基组选用;判断MCSCF的波函数的稳定性的方法;不同多参考方法对活性空间要求差异;做多参考计算的常用程序盘点和使用建议;提供大量MCSCF和多参考计算的相关资料
• 使用ORCA和Gaussian程序做CASSCF和多参考计算的实例:盘点程序的MCSCF和多参考计算的相关功能;极尽详细、全面介绍ORCA和其它程序做MCSCF和各种多参考计算的功能和计算设置,以及大量相关细节(如对非收缩MRCI的CSF的selection策略);加速积分计算设置;多个态的计算方法;CASSCF的CI系数和轨道系数不收敛的解决方法。之后给出一大批非常详细、完整的CASSCF/多参考的计算例子,每个例子都有显著的差异性,并且在讲解时特别注意让学员领会活性空间选择的思想、充分体现之前讲的活性空间的选择思路,尽最大努力令学员能举一反三面对自己实际研究的情况能选择恰到好处的活性空间,做到游刃有余。并且每个例子都有详细的分析讨论(特别是查看密度矩阵、组态函数的构成和组态系数、用Multiwfn观看活性轨道),以令学员能充分领会计算结果、加深对MCSCF方法的理解。例子如下:
态平均CASSCF计算空间简并的基态碳原子、结合Multiwfn获得球对称的电子结构
对苯酚做态平均CASSCF计算得到n-π*和π-π*激发能
CASSCF和OVB-MP2计算氧气分子单-三重态能量差
CASSCF计算臭氧,包括做优化和振动分析。利用CCSD自然轨道定义活性空间
CASSCF计算C4H8双自由基,包括计算双激发特征。利用UNO轨道定义活性空间
乙醇C-O键断裂过程的扫描的CASSCF研究。利用成键和反键的定域化的轨道定义活性空间
乙烯二面角旋转的CASSCF的过渡态搜索、振动分析和势垒的计算。利用高自旋态的SOMO轨道定义活性空间
HNO2的CASSCF计算。此例利用MP2弛豫密度的自然轨道定义活性空间
D8h对称性的处于一阶鞍点的环辛四烯的CASSCF优化和振动分析
CASSCF计算Inorg. Chem., 62, 19986 (2023)(介绍见http://sobereva.com/696)里首次研究的电子结构特殊的B6C6N6环状分子,将有限温度DFT计算得到的轨道作为活性轨道。并通过此例对比不同加速计算策略
CASSCF做乙烯二面角扭转的刚性势能面扫描,获得S0&S1&T1&态平均能量的势能曲线图
NEVPT2做N2的高精度键解离势能面扫描
NEVPT2计算呋喃的垂直激发能,强调对不可约表示的考察和考虑
CASSCF优化1,4-脱氢苯的双自由基态,强调对活性轨道做定域化的意义
MC-RPA和SA-CASSCF计算戊搭烯的π-π*激发能
标量X2C-NEVPT2对Cr2做势能面扫描得到结合能曲线
SC-NEVPT2计算吖嗪的反转S1-T1能隙
NEVPT2计算Diels-Alder反应势垒和反应能的例子和活性空间选取的详细讨论
QD-NEVPT2的计算LiF的最低两个1∑+态曲线考察避免交叉的例子和结果的深入讨论
MRCI计算CH2的最低单/三重态能量随键角的变化
MRCI+Q计算CH2的特定不可约表示的态的能量随键角的变化
MRCI+Q计算笑气NNO的异构化能并结合此例对计算异构化能所用的活性空间问题做深刻的讨论
NEVPT4(SD)计算乙酸-Cu双核配合物的单-三重态能量差
MRCC计算静态相关极强的H4体系
MRCI、FIC-MRCC、MR-EOM-CC计算Al原子的3p-4s激发能
FIC-CASPT2-IPEA做N2势能曲线扫描
借助AVAS初猜轨道做二苯甲烷的π-π*激发的NEVPT2计算
借助AVAS初猜轨道做顺铂的d-d跃迁的MRCI计算并体现double-shell的考虑
• 巨大活性空间的近似求解方法:活性空间尺寸与组态函数(CSF)和行列式数目的关系;Full CI的近似求解:Selected CI (SCI);CASSCF的近似求解策略;SCI-SCF类方法ASCI-SCF和ICE-SCF方法的具体介绍;简介其它实现大活性空间CASSCF的方法HCISCF、DMRG-SCF、FCIQMC-SCF、iCISCF、v2RDM-CASSCF。给出用ORCA做ICE-CI近似实现LiH的Full CI的例子;ORCA做ICE-SCF计算五并苯(用到CAS(22,22)活性空间)的例子;ORCA做ICE-NEVPT2结合CAS(18,18)计算18轮烯的例子
• 使用Dalton在CASSCF下做磁属性计算:Dalton是唯一能在CASSCF级别下做磁属性计算的程序,因此有独特意义,在本节介绍。本节示例用Dalton对苯分子在CASSCF级别下计算磁化率和原子的磁屏蔽值并顺带讲解怎么做NEVPT2计算,还示例对C16碳环的三重态在CASSCF级别下计算NICS(1)ZZ芳香性指数(重复卢老师在《全面揭示16碳环(cyclo[16]carbon)非常奇特的激发态芳香性!》http://sobereva.com/741介绍的论文中的计算) - 势能面交叉与光化学(130多页):不同电子态的势能面在特定几何结构下可以出现相交或接近相交,称为势能面交叉,这与光化学机理、电子激发态的无辐退激过程、涉及多态的反应的产物分布等方面有直接联系,是做非绝热过程研究的关键。本节的目的是让学员充分具备研究这些问题的能力,给出了涵盖各种情况的大量例子使得学员能够很容易地举一反三,并充分领会实际计算中的各种重要细节和关键要点。本节有三部分内容:
• (1) 势能面交叉的相关知识:这部分内容目的是使得学员对势能面交叉及各种相关背景知识有充分、深刻的了解,讲授内容包括:势能面交叉的概念;绝热态与透热态;势能面完全相交的条件与避免交叉;势能面交叉区域的势能面结构、圆锥交叉;MECP与MECI的概念;MECP的搜索算法;MECI的搜索算法;不同理论方法描述势能面交叉的能力;光化学的概念;大量的扩展阅读文章;势能面交叉与光化学的相关专著盘点
• (2) MECP搜索例子:这部分详细示例和讲解怎么用具体程序以各种方式搜索各种情况的MECP,包含以下内容
• Gaussian用户搜索MECP的方法说明
• sobMECP的用法介绍(sobMECP是本培训讲授者卢天开发的被广泛使用的搜索MECP的程序)
• 用sobMECP在DFT下搜索FeO+的四-六重态MECP实例
• 用sobMECP在DFT下搜索Pt配合物的单-三重态MECP实例
• 用sobMECP在DFT下搜索O3的S0-T1的MECP实例
• 用sobMECP在DFT下搜索氮宾分子环化的MECP实例
• 用sobMECP在TDDFT/DFT下搜索乙酰苯的S1-T1态的MECP实例
• 用Gaussian在CASSCF下搜索CH2的T0-S1的MECP实例
• ORCA搜索MECP的功能的介绍
• ORCA在DFT下搜索[CH3O]+的单-三重态的MECP实例
• ORCA在DFT下搜索乙烯二酮解离路径的MECP实例
• (3) MECI搜索例子:这部分详细示例和讲解怎么用具体程序以各种方式搜索各种情况的MECI,包含以下内容
• Gaussian用CASSCF搜索1,3-丁二烯的S1-S2的MECI实例
• Gaussian用CASSCF搜索苯-盆苯之间光异构化的MECI实例
• Gaussian用CASSCF搜索光诱导噻吩开环的MECI实例
• ORCA搜索MECI的功能的介绍
• ORCA用SF-TDDFT搜索亚乙基乙叉(ethylidene)的S0-S1间MECI实例
• ORCA用SF-TDDFT搜索亚乙基乙叉的S0-T1间的MECP实例
• ORCA用SF-TDDFT搜索偶氮苯的光异构化的MECI实例
• ORCA用SF-TDDFT搜索二苯乙烯的光环化的MECI实例
• ORCA用TDDFT搜索尿嘧啶的S1-S2间的MECI实例
• GAMESS-US搜索MECI的功能介绍
• GAMESS-US用MRSF-TDDFT搜索乙烯结构扭曲的S0-S1间MECI实例 - DFTB、GFN系列方法与xtb程序的使用(约130页):这一节重点讲愈发流行、重要性越来越高的半经验级别的DFT方法GFN-xTB以及相关方法的原理、应用,并给出如今研究大体系几乎离不开的xtb程序做GFN系列方法计算的实际例子。内容:
• DFTB系列方法:DFTB概况;DFTB的历史(DFTB的1、2、3代);DFTB的方法特点;支持DFTB计算的程序盘点;DFTB的Slater-Koster参数文件的获得和选择;量子化学程序中做SCC-DFTB计算的关键词和计算胡萝卜素的实际例子;TD-DFTB计算激发态的特点、速度和精度;量子化学程序中做TD-DFTB算尿嘧啶激发能以及胡萝卜素UV-Vis谱的例子以及结果与ZINDO和TD-PBE0的对比
• GFN系列方法:GFN系列方法的历史;GFN1-xTB的原理和特点;GFN2-xTB;自旋极化的GFN-xTB(spGFN-xTB);GFN0-xTB;GFN-FF以及用于周期性的变体pGFN-FF和mcGFN-FF;不同GFN方法之间的差异梳理;全面展示GFN系列方法算不同问题的精度(构象能量差、相互作用能、蛋白质结构优化、过渡金属配合物结构、热力学校正量等);GFN方法与GBSA/ALPB/CPCM-X溶剂模型的结合;GFN系列方法的扩展应用;精度比GFN-xTB高一个档次的g-xTB方法介绍;极快速产生不错质量波函数的PTB方法;支持GFN系列方法的程序盘点
• 做GFN系列方法计算的关键程序xtb的使用:xtb计算耗时与并行核数的关系;xtb的安装;xtb的运行命令;整体计算控制选项;控制计算方法的选项;控制任务类型的关键选项;其它任务类型的选项;溶剂模型设置;输出控制;控制文件的详细设置方法;xtb的输出文件;GFN2-xTB做气相和溶剂下的OPP双环分子的单点计算的例子、输出信息解读以及用Multiwfn看轨道;GFN2-xTB和GFN-FF做富勒烯嵌入OPP分子形成的复合物的结构优化的例子(注:此例和《8字形双环分子对18碳环的独特吸附行为的量子化学、波函数分析与分子动力学研究》http://sobereva.com/674介绍的卢老师的研究工作密切相关);GFN-FF优化COF晶胞的例子(其中利用Multiwfn转换晶体文件格式);GFN-FF对288原子的[144]-轮烯做极快速振动分析的例子,以及用Multiwfn绘制红外光谱,顺带讲解对xtb消虚频的方法;xtb做势能面扫描的例子;xtb结合Double-Ended Growing String Method (DE-GSM)方法产生反应路径的介绍和将此方法用于环丙基卡宾异构化的实例;通过Gaussian调用xtb(利用卢老师写的gau_xtb脚本,见http://sobereva.com/421)以及通过这个组合研究Sc引发的C-H活化过程的例子;通过ORCA调用xtb;ORCA 6.1开始加入的原生的GFN-xTB功能用法;用xtb程序将极化率分解为原子的贡献(注:更准确的方法是《使用Multiwfn计算分子中的原子极化率》http://sobereva.com/600介绍的做法)
- 从头算分子动力学(240多页):从头算动力学(AIMD)是极其强大的模拟技术,相对于普通力场的分子动力学来说,AIMD不仅能做到明显更精确、几乎可以直接用于任意体系(而不需要考虑准备合适力场参数、构建拓扑信息的麻烦),还可以研究动力学中涉及成键和断键的过程,如热解、异构化、化学反应等。这一节超级完整全面讲解从头算动力学的知识,给了巨量十分精心设计的例子,充分体现AIMD研究需要掌握的方方面面细节,还介绍了大量高阶经验技巧,可以令对从头算动力学感兴趣的学员们一次学个痛快,而且感到十分有趣!这部分是高级班的关键主题之一,内容如下。注意:本节假定学员已经具备基于经典力场的分子动力学模拟技术的相关知识,如热浴的原理、动力学走步算法和步长的选择等,也假定学员已会用VMD、会tcl分析脚本的编写,不具备这些知识者强烈建议参加北京科音分子动力学与GROMACS培训班(http://www.keinsci.com/KGMX)学习相关部分。本节只讲孤立体系的从头算动力学模拟,如果要对周期性体系做这种模拟,应当参加北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)学习使用CP2K来做。另外,本节不讲授路径积分分子动力学(PIMD),这部分在北京科音CP2K第一性原理计算培训班里有专门的一节详细讲授相关知识以及用CP2K做PIMD的方法。
• 从头算动力学的基本知识:什么是从头算分子动力学?、经典(classical)的含义详解、AIMD的三种形式(BOMD、CPMD、ADMP)、CPMD的基本思想、AIMD的相关学习资料、可以做AIMD的程序、AIMD程序的选择建议、做孤立体系AIMD常用的三种程序的对比、准备可视化程序VMD
• 使用xtb程序做AIMD:运行方法和关键词、模拟要点、输出文件说明、耗时与并行效率测试
实例1:1,2,4,5-Tetrazine(均四嗪)热分解
实例2:He原子在螺烯上的吸附行为。这部分重复《谈谈范德华势以及在Multiwfn中的计算、分析和绘制》(http://sobereva.com/551)介绍的卢老师提出的范德华势的原文里的动力学模拟体系
实例3:考察柔性药物分子瑞德西韦的动力学行为。这部分重复《使用Molclus结合xtb做的动力学模拟对瑞德西韦(Remdesivir)做构象搜索》(http://bbs.keinsci.com/thread-16255-1-1.html)里所介绍的模拟
实例4:富勒烯形成机理的动力学研究。重复Theory and Applications of Computational Chemistry: The First Forty Years第31章里的模拟,由较短的碳纳米管自发封端变成富勒烯,以及大量C2分子在纳米反应器里自发变成富勒烯
实例5:氧气分子与石墨烯空位反应。这部分完美重现J. Phys. Chem. C, 122, 29368 (2018)文中的研究结果
• 使用ORCA程序做AIMD:AIMD模块的功能和特征、单位、控制动力学的$md字段命令的特别详细完整的介绍和解释、模拟相关问题和细节、跑AIMD推荐的计算级别、模拟速度与并行核数的关系
实例1:18碳环的动力学行为。重复《揭示各种新奇的碳环体系的振动特征》(http://sobereva.com/578)中介绍的卢老师的18碳环振动行为的研究工作
实例2:β-propiolactam(丙内酰胺)的热解过程的模拟
实例3:(HCOO-)(H2SO4)阴离子团簇内的质子转移的模拟。此例重复J. Am. Chem. Soc., 139, 11321 (2017)中的模拟体系,并给出VMD tcl脚本对不同方式质子转移对应的不同构型分布比例进行统计分析
实例4:乙炔的燃烧过程。此例模拟O2以恰到好处的朝向高速与乙炔碰撞导致产生H自由基、HCO自由基和CO分子,并且通过shell脚本利用波函数分析程序Multiwfn对整个模拟轨迹的自旋布居、键级、LOL的变化进行直观的考察,从而对电子结构的变化提供非常丰富的信息,大大增加了研究深度!
实例5:18氮环分子解离过程的模拟。此例重复《18个氮原子组成的环状分子长什么样?一篇文章全面揭示18氮环的特征!》(http://sobereva.com/725)介绍的卢老师的AIMD研究。结合此例专门强调计算级别的恰当选择,以及温度对解离速率、产物的影响。
练习:[Al(H2O)6]3+与NH3之间的质子转移
metadynamics模拟:这个部分介绍如今非常热门的metadynamics (MTD)模拟的基础知识,以及通过ORCA实现MTD的方法和实际模拟例子
• 使用Gaussian做AIMD:讲解Gaussian的AIMD功能的概况、ADMP模块的使用、BOMD模块的使用、模拟方式以及模拟初始条件的设置(这对于做准经典动力学是重中之重的关键点!)、将AIMD轨迹转换为xyz格式、ADMP与BOMD的并行效率问题
实例1:草酸的动力学模拟、热解的模拟。此例演示了ADMP的使用、输出信息的正确理解、控温的方式,以及怎么用BOMD做准经典动力学
实例2:F- + CH3Cl → CH3F + Cl-的SN2反应的动力学模拟。此例演示如何恰当设置初速度令期望的反应发生,以及对反应过程的几何信息进行分析。同时还结合此例讲解direct和indirect反应方式、无反应碰撞、微溶剂产生的影响
实例3:甲醛的光解离。此例研究甲醛从S1态的2141振动态内转换回S0基态后由于具有很高动能而克服势垒发生光解离的过程。结合此例具体讲解了怎么设置判断解离的标准、恰当设定虚频模式的初始动能
实例4:水合Fe(II)的动力学模拟
实例5:H2DCO异构化中势能面的二分研究。在北京科音的中级量子化学培训班的IRC部分中专门讲过势能面的二分,这个实例通过从过渡态出发的50次准经典AIMD模拟得到了可靠的经历势能面二分的异构化反应分支比
• 其它相关知识:做AIMD的Progdyn和VENUS程序简介、AIMD在理论预测质谱中的应用的详细介绍、通过AIMD计算振动光谱、量子动力学简介。注意路径积分分子动力学(PIMD)在北京科音CP2K第一性原理计算培训班(http://www.keinsci.com/KFP)专门有一节详细讲授并给了CP2K实现的具体例子,因此不在高级量子化学培训班里展开讲。 - real-time TDDFT模拟(40多页):real-time TDDFT (RT-TDDFT)通过求解含时Kohn-Sham方程,可以模拟体系的能量和电子结构特征随时间的实时变化。常用于模拟随时间变化的电场对化学体系的能量和电子结构的影响、模拟价层和X光电子光谱、考察电子转移等超快过程。本节详细介绍RT-TDDFT的各种相关知识以及通过NWChem做RT-TDDFT模拟的方法,内容包括:RT-TDDFT的原理和用途介绍;支持RT-TDDFT的程序盘点;RT-TDDFT的相关学习资料;使用NWChem做RT-TDDFT的输入文件写法详解;做RT-TDDFT模拟电子光谱的流程和细节的详细介绍;
实例1:施加kick场模拟水分子的全范围电子光谱并与LR-TDDFT的结果进行对比,顺带强调RT-TDDFT具有的模拟吸收光谱在强场下表现的非线型特征的能力、强调RT-TDDFT算大体系完整光谱相对于LR-TDDFT的效率优势
实例2:模拟高斯包络脉冲电场令水分子产生的共振激发
实例3:模拟外加的电子在TCNE二聚体中的分子间瞬时转移过程 - sTDA方法快速计算电子光谱(36页):这一节讲授能超级快速计算很大体系电子光谱的sTDA方法及其各种衍生方法,sTDA-xTB甚至能计算含几千个原子的蛋白质的电子光谱!内容包括:sTDA-DFT和sTDDFT的原理、特点、精度、耗时、截断阈值等参数的设置;sTDA-xTB和sTD-xTB方法的原理和各种实际应用(不限于电子光谱,还有光学旋转、SHG、HRS、TPA等);支持sTDA的程序盘点;XsTD/XsTDA-DFT方法;使用ORCA做sTDA/sTD计算的方法;sTD-DFT计算十二苯基并四苯的例子、结合Multiwfn绘制UV-Vis和ECD光谱,以及与昂贵得多的TDDFT的对比;使用xtb4stda结合stda程序做sTDA-xTB计算的方法,以及用于十二苯基并四苯的例子(150个原子算100多个激发态,仅仅约10秒钟算完!)
- 振动电子光谱的计算(91页):实际中振动与电子跃迁是存在耦合的,对应的光谱叫做振动电子光谱,需要在计算电子光谱时考虑振动耦合效应。然而大多数计算电子光谱的人忽略了振动耦合,此时得到的光谱缺乏精细结构,往往因此和实验峰型对比得不理想。此外,对称禁阻的电子跃迁仅在考虑振动耦合时才能发生,并且振动耦合使得很多振子强度很小的电子跃迁在实际中不那么微弱。可见懂得如何在计算电子光谱中考虑振动耦合对于专业研究分子光学特征的人来说有极为重要的意义。这一节特别完整详细讲授振动电子光谱的各种背景知识和实际计算方法,内容包括三大部分:
• 振动电子光谱的理论知识和计算方法:振动电子光谱的概念;考虑振动耦合时的跃迁电偶极矩的表达式、FC积分和FC因子;Franck-Condon (FC)原理;初、末态正则坐标间的变换关系、Duschinsky矩阵J、位移矢量K;Herzberg-Teller (HT)效应;温度对振动电子光谱的影响;不同振动电子光谱的计算模型(Adiabatic Hessian、Vertical gradient、Adiabatic Hessian After a Step、Vertical Hessian、Adiabatic Shift)的方法和对比;SOS(亦称TI)方法计算振动电子光谱曲线(原理、class的含义、初筛方式、convergence判断、low progression问题的解决);路径积分(亦称TD)方法计算振动电子光谱曲线的原理和相对于SOS的优缺点;振动电子光谱的非谐振校正;Huang-Rhys因子的定义和与重组能的关系;提供大量相关资料作为学员的扩展阅读
• 使用Gaussian计算振动电子光谱:丙烯醛的振动分辨的吸收、发射光谱计算的完整实例(各种相关设置全面的说明、关键词的写法、输出信息解读、图像绘制、峰的指认、FC和FCHT结果的对比、考虑温度效应的效果,输出J矩阵、K矢量和Huang-Rhys因子,分析电子结构改变导致的结构变化与振动模式之间的关系);计算(Z)-8-Methoxy-4-Cyclooctenone (MCO)的n-π*的振动分辨吸收光谱的实例,结合此例讲解遇到low progression时忽略个别振动模式解决问题的重要方法,并与文献里的结果对比;计算SF6-的光电离谱的实例,通过此例强调恰当调节阈值参数的重要性
• 使用ORCA计算振动电子光谱:介绍ORCA的计算振动电子光谱的功能,并且给出ORCA对丙烯醛做振动电子光谱计算的完整例子,包括振动分辨吸收光谱(分别使用AH、VG、AHAS方式计算并对比结果令学员直观了解它们的差异并给出使用建议。还讲解同时考虑多个激发态的这种光谱的绘制)、振动分辨荧光光谱、振动分辨ECD光谱、振动分辨CPL光谱。同时还讲解考虑振动耦合影响时的荧光速率的精确计算方法。 - 磷光速率的计算(93页):磷光是化学体系的重要的发光方式,通常对应T1态到S0态的辐射过程,磷光速率和磷光寿命是研究分子发光问题的人极为关注的,本节一次性令学员充分学透这方面的知识和实际计算。本节的内容:
• 磷光速率的概念与计算方法:介绍磷光的概念;磷光量子产率;磷光速率如何由三重态的各子态的激发能、跃迁偶极矩/振子强度、Boltzmann分布得到;盘点磷光速率的计算方法(微扰法、变分法、二/四分量相对论、二次响应理论);详细介绍微扰法获得T1-S0间跃迁电偶极矩的原理;详细介绍响应理论的各种相关知识,特别是响应函数与跃迁属性之间的关系,这对于理解Dalton程序做响应理论计算的原理至关重要。
• 使用Dalton计算磷光速率和旋轨耦合矩阵元:讲解使用Dalton通过响应函数方式计算磷光速率的具体方式、此程序与之相关的功能和特征。然后给出计算2-硫代胸腺嘧啶磷光速率的例子(涵盖各种情况,包括完整旋轨耦合积分、AMFI方式、Zeff方式算旋轨耦合,以及计算时考虑PCM溶剂模型和DKH2标量相对论效应的做法、使用ANO类基组的方法)。接下来给出含有过渡金属的较大配合物Ir(bzq)2(acac)算磷光速率的例子并与实验值进行对比。之后顺带对很多人关心的Dalton计算旋轨耦合矩阵元的做法进行详细讲解,包括线性响应函数的残值方式计算S0和不同三重态激发态间的旋轨耦合矩阵元,以及二次响应函数的双残值方式计算不同激发态之间的旋轨耦合矩阵元。
• 使用ORCA计算磷光速率和绘制振动分辨磷光光谱:首先讲解通过SOC-TDDFT结合变分法计算配合物Ir(bzq)2(acac)磷光速率的方法和要领。然后讲解考虑振动耦合计算联乙酰的磷光速率,这部分利用了ORCA的ESD模块给出考虑了Franck-Condon、Herzberg-Teller效应以及温度效应的磷光速率,并同时讲解得到准确的振动分辨磷光发射谱的方法,并与文献报道的此体系的室温磷光发射谱进行对比。
• 使用Dirac计算磷光速率:这部分介绍怎么使用Dirac程序做二分量相对论TDDFT计算直接算磷光速率。考虑到学员几乎都没用过专门擅长做二/四分量相对论计算的Dirac,首先对Dirac程序的功能进行介绍,然后讲解Dirac的安装,介绍程序的运行常识和输入文件的结构。之后讲解Dirac结合X2C二分量哈密顿用TD-B3LYP/dyall.3zp级别算H2CS分子的方法,并说明如何通过输出信息得到准确的磷光速率。然后讲解以类似方式对Ir(bzq)2(acac)配合物算磷光速率的例子,并强调在收敛方面要注意的要点。
- 内转换与系间窜越速率的计算(40多页):内转换(IC)和系间窜越(ISC)是光化学、发光材料等方面研究极其关注的内容,掌握IC和ISC速率的计算能使得这方面的研究深度极大地加深。这一节有两大部分内容:
• 内转换与系间窜越的基本知识及速率的计算方法:介绍内转换和系间窜越的相关概念;介绍重要的判断ISC可能性的El-Sayed's规则;列举系间窜越速率的估计/计算方法(靠能量最粗略估计的方式、结合旋轨耦合估计、定量计算,说明Herzberg-Teller (HT)效应的重要性,并提及计算的一些关键细节);Fermi's黄金规则、Marcus理论、Landau-Zener理论计算ISC速率的具体公式讲解;内转换速率的估计/计算方法;Fermi's黄金规则和Marcus理论计算IC速率的具体公式讲解;全面盘点计算IC速率所需要的非绝热耦合矩阵元(NACME)的各种程序
• 使用ORCA计算内转换和系间窜越速率:详细介绍ORCA计算IC和ISC的功能;对乙酰苯计算ISC速率给出极其详细完整的例子,把每一步的各方面计算细节都讲得十分透彻以确保学员能够成功复现例子并举一反三成功计算其它体系,流程具体包括对S0做优化+振动分析、对S1做优化+振动分析并估计要考察的T态、对T1做优化+振动分析+SOCME计算、不考虑HT的方式计算ISC速率、考虑HT的方式计算ISC速率;给出计算蒽的ISC速率的练习(提供了全套输入文件供自行对照);给出计算薁(azulene)的S1-S0内转换速率的完整例子并讲解各方面细节和注意事项
- GAMESS-US的使用基础(近90页):通过这一节学员可以快速上手很知名、开源免费的GAMESS-US程序,此程序在后面几节需要利用到(双光子吸收、高级振动分析、(MR)SF-TDDFT部分)
• GAMESS-US的概况:功能、优点、缺点、历史与分支、GAMESS-US的手册、御用的可视化程序wxMacMolPlt
• GAMESS-US的安装和配置:Linux版的编译、配置运行环境、测试、使用方法;Windows版的安装和运行
• GAMESS-US的输入文件:输入文件的基本规则、内存的设置、$CONTRL字段、$DATA字段、关于对称性的利用、基组的指定、赝势的设置、溶剂模型($PCM)、$GUESS字段、几个重要关键词、利用Gaussian产生坐标和基组定义、用Multiwfn产生GAMESS-US输入文件、产生波函数文件、SCF不收敛的解决
• GAMESS-US的简单任务示例:气相下ωB97XD/pcSeg-1算卟啉单点(包括利用wxMacMolPlt设置对称性、看轨道);内坐标下用HF/3-21G*计算SH6;乙醇溶剂下用B3LYP/6-31G*优化甲醛;真空下PBE0/6-311G**对甲醛优化和做振动分析;BP86-D3(BJ)/SDD&TZVP计算顺铂单点能;BP86-D3(BJ)/SBKJC&6-311G*计算顺铂单点能;UMP2/cc-pVTZ计算三重态乙醇的受力;TDDFT PBE0/6-31G*对靛青算最低10个单重态激发态;ROCCSD/cc-pVTZ计算CH自由基;CCSD(T)/cc-pVDZ扫描甲酰胺C-N键长;G4(MP2)-6X热力学组合方法计算LiH;GAMESS-US中的CI计算方式说明、CISD/cc-pVTZ计算乙炔 - 双光子吸收的计算(24页):双光子吸收是化学体系同时吸收两个光子到达激发态的过程,是分子光学重要的组成部分。这一节使得学员了解相关知识并学会怎么理论计算,内容有:双光子吸收(TPA)的基本概念;TPA截面、TPA强度以及二者的转化关系的详细说明;TPA的计算方法(SOS和响应函数);计算TPA的理论方法和基组;支持TPA计算的程序盘点;使用GAMESS-US基于TDDFT计算有机硼化合物的TPA的具体例子以及和文献的对比;使用Dalton基于TDDFT计算TPA详解;使用Dalton基于耦合簇高精度计算TPA的例子。还同时介绍三光子吸收并给出Dalton做三光子计算的实例
- spin-flip TDDFT方法(66页):本节详细介绍目前已用得很广泛的spin-flip TDDFT的原理和应用,对于研究单/双/多自由基的基态和低阶激发态的能量、结构、交叉非常有用,比多参考计算容易、便宜得多。内容有:
• spin-flip的相关知识:spin-flip (SF)方法的思想;以CH2卡宾计算为例解释SF计算牵扯的行列式和结果;SF算三自由基(triradical)的情况;SF用于乙烯二面角扭转的研究;SF-TDDFT的形式和泛函选择;SF-TDDFT的实际用途;SF的相关文章;支持SF的程序盘点;SF-TDDFT的改进方法MRSF-TDDFT原理和实际用处的详细介绍以及它相对于SF-TDDFT的优点的讨论,并简要介绍专长做MRSF-TDDFT的OpenQP程序
• 在GAMESS-US和ORCA中做(MR)SF-TDDFT计算的例子:此类计算的功能和设置方法;在CH2的基态结构下做SF-TDDFT计算的实例;SF-TDDFT计算长烃链卡宾的电子态能量差、激发态波函数的实例;SF-TDDFT优化m-benzyl双自由基的基态实例;用SF-TDDFT计算1,3,5-脱氢苯三自由基的二重态基态与四重态的绝热能量差的实例;GAMESS-US里做MRSF-TDDFT的设置方法,给出此方法计算1,3-丁二烯最低5个激发态的详细例子,并结合此例强调它描述双激发特征显著的态和势能面交叉方面的能力 - 分子振动问题的高级计算方法(60页):本节深入讲授振动分析的底层知识以及怎么非常精确地计算分子振动问题。如果想极高精度地计算振动光谱,这一节是必学的。首先讲常用的谐振近似处理振动问题的局限性、考虑非谐振效应的三种常见方式,然后对振动二阶微扰理论(VPT2)和振动自洽场(VSCF)及相关方法两大类方法分别展开讲解。
• VPT2部分:VPT2的思想;含三模式耦合的四次力场;VPT2的基频、泛音、合频的表达式;VPT2方法的变体(GVPT2、DCPT2、HDCPT2);VPT2非谐振校正个别情况异常大的问题;VPT2方法的耗时;支持VPT2方法的程序盘点(Gaussian、ORCA、CFOUR、Q-Chem、iGVPT、pyVPT2);杂化方式做VPT2计算;通过机器学习(ML)势节约VPT2的耗时;使用ORCA做GVPT2计算的方法以及具体计算例子和输出信息解读;使用ORCA获得振动校正后的NMR化学位移(对于高精度计算NMR很重要);ORCA做廉价的非谐振计算(NEARIR关键词)
• VSCF及相关方法部分:VSCF的特点;VSCF的原理的深入介绍;2MR-QFF和direct方式做VSCF;VSCF用的坐标;在VSCF基础上的改进,包括VSCF-PT2(亦称cc-VSCF或VMP2)的原理和相对于VPT2的优缺点、VSCF-CI、VCC、VMCSCF、VMRCI;做VSCF及衍生方法的计算级别的选择;使用GAMESS-US做VSCF及相关计算,包括$VSCF中的各种关键设置详解,甲醛的VSCF-PT2、VCI、VDPT2的计算实例讲解和输出信息的非常详细的说明 - X光光谱的计算:量子化学研究者研究的最多的电子光谱的是价层电子激发相关的光谱,而内核电子的激发(对应X光吸收光谱,XAS)和内核电子的电离(对应X光电子谱,XPS)也同样十分重要,能够提供原子所处状态、局部化学环境方面的关键信息。本节讲授X光光谱的各种相关背景知识,详细盘点各种计算X光光谱的方法并进行比较(如TDDFT、STEOM-CCSD、IP-EOM-CCSD、CASCI/NEVPT2、DFT/ROCIS、结合MOM做ΔSCF、Koopmans定理结合QTP系列泛函等),并给出以不同方法计算各种体系的XAS和XPS的实际例子,使研究者能够举一反三、轻车熟路地做这类计算
- 对角Born-Oppenheimer校正(DBOC)的计算(12页):DBOC是计算极高精度计算热化学数据(达到亚化学精度)必须考虑的一项。此节详细介绍了DBOC的概念、重要性和计算的背景知识,并演示怎么用ORCA和CFOUR程序分别在DFT和耦合簇级别下计算DBOC
- 转动常数的计算(11页):分子转动光谱是分子光谱学中重要的组成部分,与分子的转动常数密切相关。此节讲怎么严格、准确地计算转动常数。内容:转动常数的定义;双原子转动能级公式;转动常数的振动校正项的概念和计算;计算二环[2.2.2]辛二烯的转动常数的完整实例、输出信息解读、与实验值的对比